Bioinformatics Interdepartmental Program, University of California, Los Angeles, CA, USA.
Department of Biological Chemistry, University of California, Los Angeles, CA, USA.
Commun Biol. 2021 Jun 3;4(1):698. doi: 10.1038/s42003-021-02231-w.
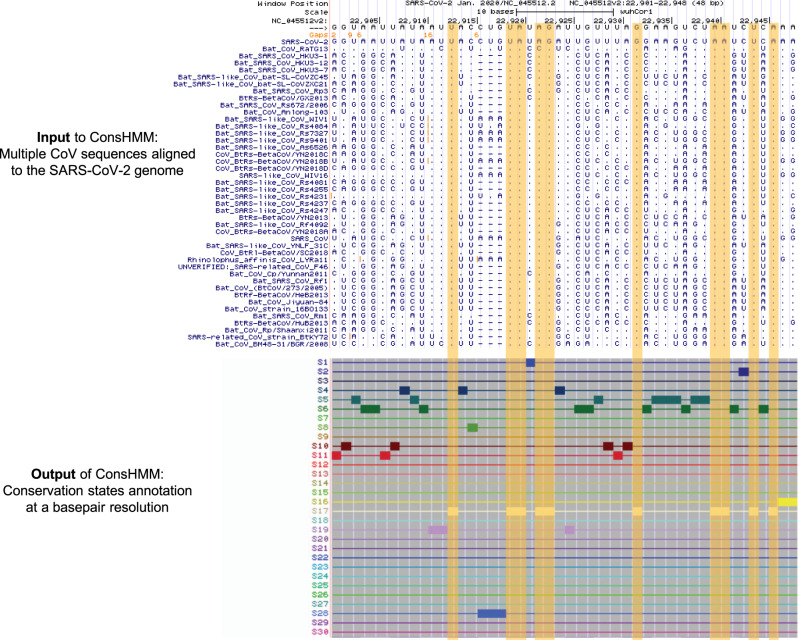
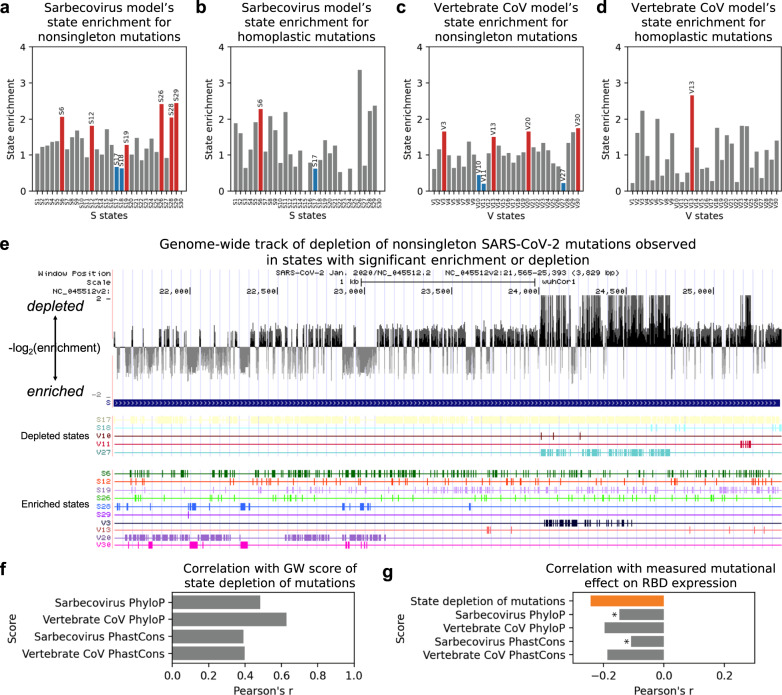
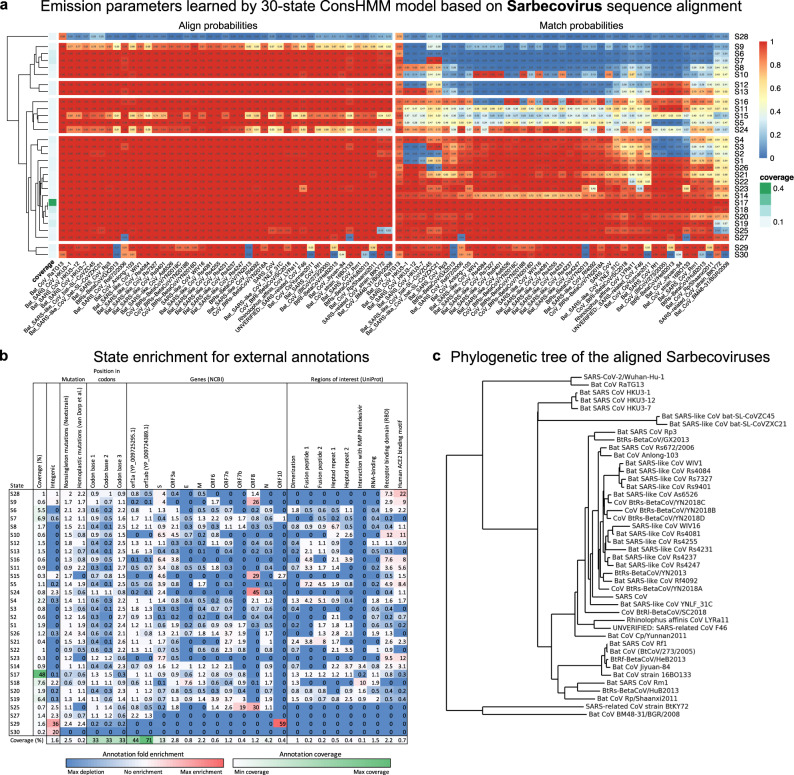
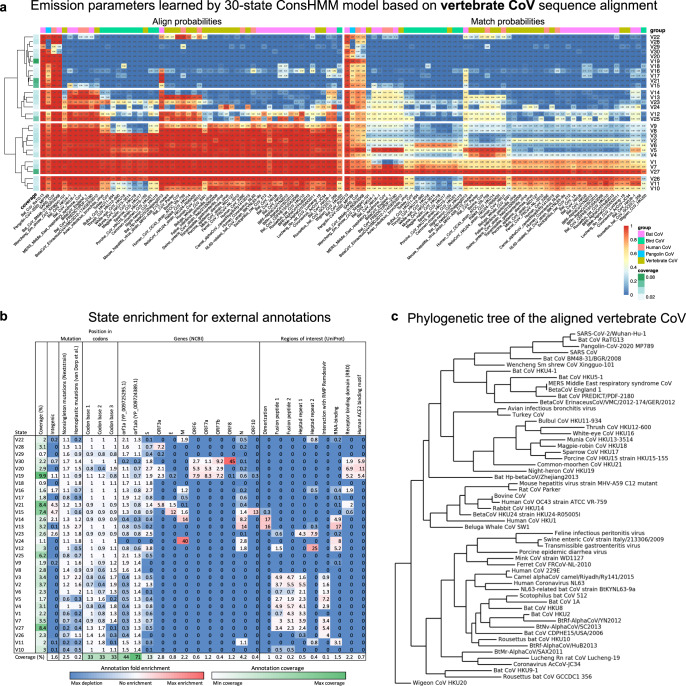
Given the global impact and severity of COVID-19, there is a pressing need for a better understanding of the SARS-CoV-2 genome and mutations. Multi-strain sequence alignments of coronaviruses (CoV) provide important information for interpreting the genome and its variation. We apply a comparative genomics method, ConsHMM, to the multi-strain alignments of CoV to annotate every base of the SARS-CoV-2 genome with conservation states based on sequence alignment patterns among CoV. The learned conservation states show distinct enrichment patterns for genes, protein domains, and other regions of interest. Certain states are strongly enriched or depleted of SARS-CoV-2 mutations, which can be used to predict potentially consequential mutations. We expect the conservation states to be a resource for interpreting the SARS-CoV-2 genome and mutations.
鉴于 COVID-19 的全球影响和严重性,我们迫切需要更好地了解 SARS-CoV-2 基因组和突变。冠状病毒(CoV)的多株序列比对为解释基因组及其变异提供了重要信息。我们应用一种比较基因组学方法 ConsHMM,对 CoV 的多株序列进行比对,根据 CoV 之间的序列比对模式,为 SARS-CoV-2 基因组的每个碱基标注保守状态。学习到的保守状态显示出对基因、蛋白质结构域和其他感兴趣区域的明显富集模式。某些状态强烈富集或耗尽 SARS-CoV-2 突变,可用于预测可能具有重要意义的突变。我们期望保守状态成为解释 SARS-CoV-2 基因组和突变的资源。