Audia Sylvain, Mahévas Matthieu, Nivet Martin, Ouandji Sethi, Ciudad Marion, Bonnotte Bernard
Service de Médecine Interne et Immunologie Clinique, Centre de Référence Constitutif des Cytopénies Auto-immunes de l'adulte, Centre Hospitalo-Universitaire Dijon Bourgogne, Université de Bourgogne Franche-Comté, Dijon, France.
INSERM, EFS BFC, UMR1098, Interactions Hôte-Greffon-Tumeur/Ingénierie Cellulaire et Génique, LabEx LipSTIC, Dijon, France.
Hemasphere. 2021 Jun 1;5(6):e574. doi: 10.1097/HS9.0000000000000574. eCollection 2021 Jun.
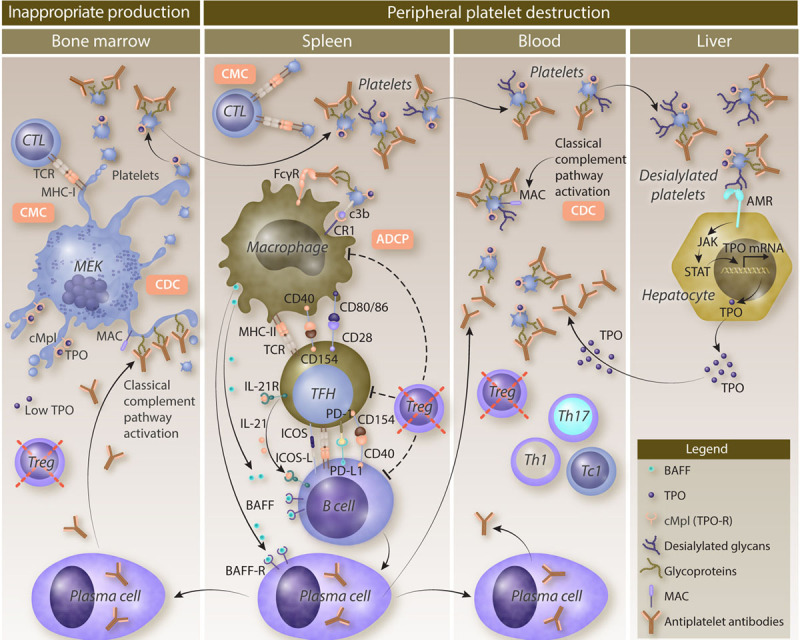
Immune thrombocytopenia (ITP) is a rare autoimmune disease due to both a peripheral destruction of platelets and an inappropriate bone marrow production. Although the primary triggering factors of ITP remain unknown, a loss of immune tolerance-mostly represented by a regulatory T-cell defect-allows T follicular helper cells to stimulate autoreactive splenic B cells that differentiate into antiplatelet antibody-producing plasma cells. Glycoprotein IIb/IIIa is the main target of antiplatelet antibodies leading to platelet phagocytosis by splenic macrophages, through interactions with Fc gamma receptors (FcγRs) and complement receptors. This allows macrophages to activate autoreactive T cells by their antigen-presenting functions. Moreover, the activation of the classical complement pathway participates to platelet opsonization and also to their destruction by complement-dependent cytotoxicity. Platelet destruction is also mediated by a FcγR-independent pathway, involving platelet desialylation that favors their binding to the Ashwell-Morell receptor and their clearance in the liver. Cytotoxic T cells also contribute to ITP pathogenesis by mediating cytotoxicity against megakaryocytes and peripheral platelets. The deficient megakaryopoiesis resulting from both the humoral and the cytotoxic immune responses is sustained by inappropriate levels of thrombopoietin, the major growth factor of megakaryocytes. The better understanding of ITP pathogenesis has provided important therapeutic advances. B cell-targeting therapies and thrombopoietin-receptor agonists (TPO-RAs) have been used for years. New emerging therapeutic strategies that inhibit FcγR signaling, the neonatal Fc receptor or the classical complement pathway, will deeply modify the management of ITP in the near future.
免疫性血小板减少症(ITP)是一种罕见的自身免疫性疾病,其病因是血小板的外周破坏和骨髓生成异常。尽管ITP的主要触发因素尚不清楚,但免疫耐受的丧失(主要表现为调节性T细胞缺陷)使滤泡辅助性T细胞能够刺激自身反应性脾B细胞,这些B细胞分化为产生抗血小板抗体的浆细胞。糖蛋白IIb/IIIa是抗血小板抗体的主要靶标,通过与Fcγ受体(FcγRs)和补体受体相互作用,导致脾巨噬细胞吞噬血小板。这使得巨噬细胞通过其抗原呈递功能激活自身反应性T细胞。此外,经典补体途径的激活参与血小板调理作用,并通过补体依赖性细胞毒性导致血小板破坏。血小板破坏也由一条不依赖FcγR的途径介导,该途径涉及血小板去唾液酸化,这有利于它们与阿什韦尔-莫雷尔受体结合并在肝脏中清除。细胞毒性T细胞也通过介导对巨核细胞和外周血小板的细胞毒性作用,参与ITP的发病机制。由体液免疫和细胞毒性免疫反应导致的巨核细胞生成不足,由血小板生成素(巨核细胞的主要生长因子)的不适当水平维持。对ITP发病机制的深入了解带来了重要的治疗进展。针对B细胞的疗法和血小板生成素受体激动剂(TPO-RAs)已经使用多年。抑制FcγR信号传导、新生儿Fc受体或经典补体途径的新兴治疗策略,将在不久的将来深刻改变ITP的治疗方式。