Department of Neurology, Fukushima Medical University, 1 Hikarigaoka, Fukushima, Fukushima, 960-1295, Japan.
Department of Otorhinolaryngology, Fukushima Medical University, 1 Hikarigaoka, Fukushima, Fukushima, 960-1295, Japan.
BMC Neurol. 2021 Jun 25;21(1):243. doi: 10.1186/s12883-021-02256-y.
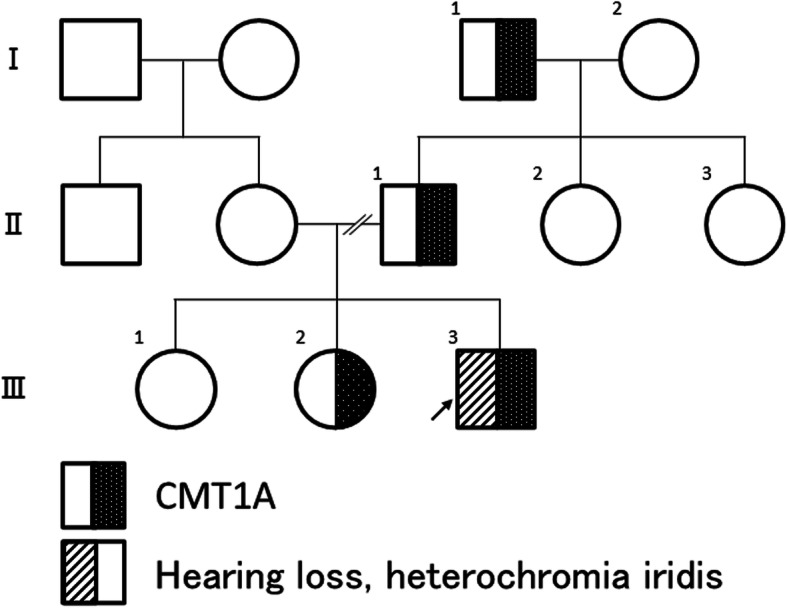
Hereditary motor and sensory neuropathy, also referred to as Charcot-Marie-Tooth disease (CMT), is most often caused by a duplication of the peripheral myelin protein 22 (PMP22) gene. This duplication causes CMT type 1A (CMT1A). CMT1A rarely occurs in combination with other hereditary neuromuscular disorders. However, such rare genetic coincidences produce a severe phenotype and have been reported in terms of "double trouble" overlapping syndrome. Waardenburg syndrome (WS) is the most common form of a hereditary syndromic deafness. It is primarily characterized by pigmentation anomalies and classified into four major phenotypes. A mutation in the SRY sex determining region Y-box 10 (SOX10) gene causes WS type 2 or 4 and peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, WS, and Hirschsprung disease. We describe a 11-year-old boy with extreme hypertrophic neuropathy because of a combination of CMT1A and WS type 2. This is the first published case on the co-occurrence of CMT1A and WS type 2.
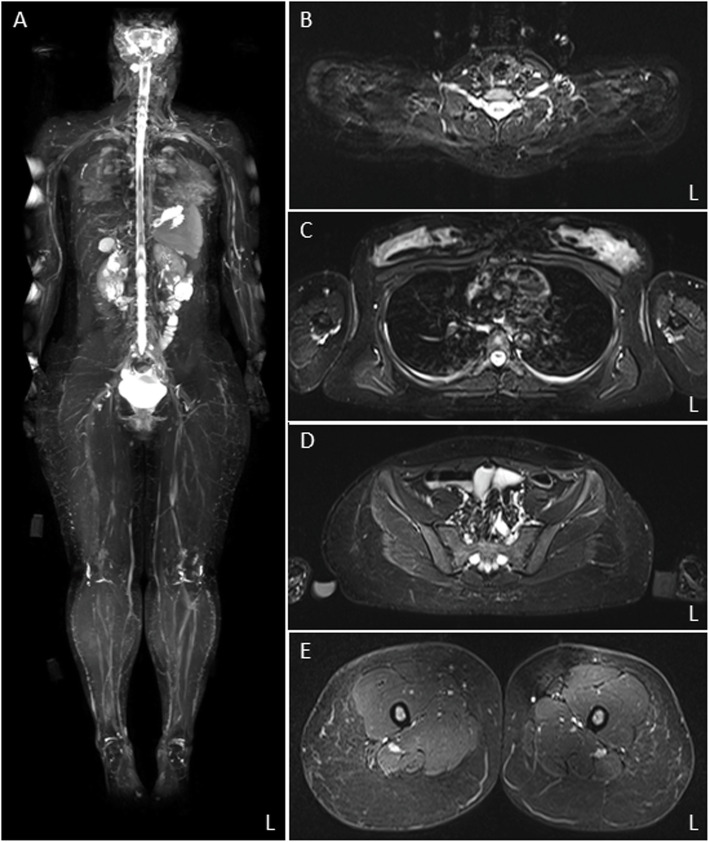
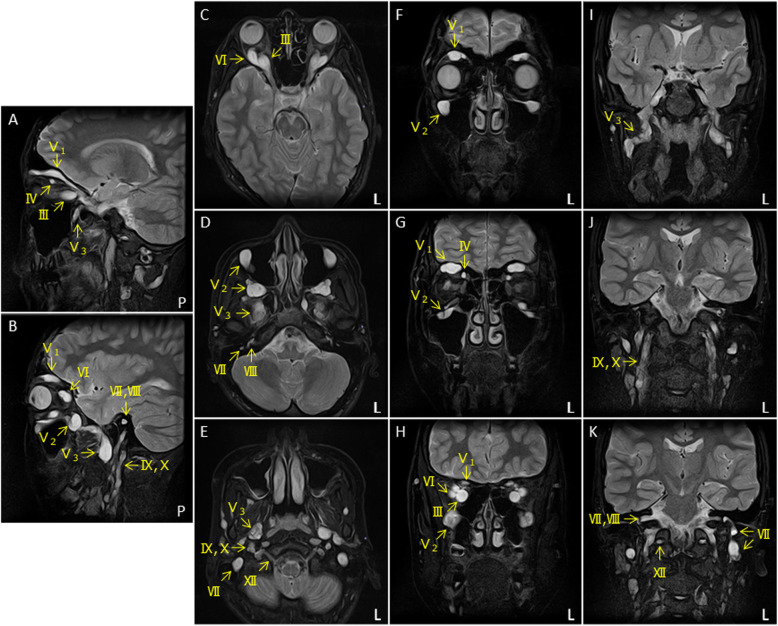
The 11-year-old boy presented with motor developmental delay and a deterioration in unstable walking at 6 years of age. In addition, he had congenital hearing loss and heterochromia iridis. The neurological examination revealed weakness in the distal limbs with pes cavus. He was diagnosed with CMT1A by the fluorescence in situ hybridization method. His paternal pedigree had a history of CMT1A. However, no family member had congenital hearing loss. His clinical manifestation was apparently severe than those of his relatives with CMT1A. In addition, a whole-body magnetic resonance neurography revealed an extreme enlargement of his systemic cranial and spinal nerves. Subsequently, a genetic analysis revealed a heterozygous frameshift mutation c.876delT (p.F292Lfs*19) in the SOX10 gene. He was eventually diagnosed with WS type 2.
We described a patient with a genetically confirmed overlapping diagnoses of CMT1A and WS type 2. The double trouble with the genes created a significant impact on the peripheral nerves system. Severe phenotype in the proband can be attributed to the cumulative effect of mutations in both PMP22 and SOX10 genes, responsible for demyelinating neuropathy.
遗传性运动感觉神经病,也称为夏科-马里-图什病(CMT),通常由外周髓鞘蛋白 22(PMP22)基因的重复引起。这种重复导致 CMT1A 型(CMT1A)。CMT1A 很少与其他遗传性神经肌肉疾病同时发生。然而,这种罕见的遗传巧合会产生严重的表型,并已在“双重麻烦”重叠综合征的报告中有所描述。瓦登堡综合征(WS)是遗传性综合征性耳聋最常见的形式。它主要表现为色素沉着异常,分为四个主要表型。SRY 性别决定区 Y 框 10(SOX10)基因突变导致 WS 型 2 或 4 型和周围脱髓鞘神经病、中枢脱髓鞘性脑白质营养不良、WS 和先天性巨结肠病。我们描述了一例 11 岁男孩因 CMT1A 和 WS 型 2 合并而患有极度肥大性神经病。这是首例关于 CMT1A 和 WS 型 2 同时发生的病例报告。
这名 11 岁男孩因 CMT1A 和 WS 型 2 合并,出现运动发育迟缓,并在 6 岁时行走不稳恶化。此外,他患有先天性听力损失和虹膜异色。神经系统检查显示远端肢体无力伴高弓足。他通过荧光原位杂交方法被诊断为 CMT1A。他的父亲家族中有 CMT1A 病史。然而,没有家族成员有先天性听力损失。他的临床表现明显比其他 CMT1A 亲属严重。此外,全身磁共振神经造影显示他的全身颅神经和脊神经明显增大。随后,基因分析显示 SOX10 基因中的杂合移码突变 c.876delT(p.F292Lfs*19)。他最终被诊断为 WS 型 2。
我们描述了一例经基因证实的 CMT1A 和 WS 型 2 重叠诊断的患者。这两个基因的双重麻烦对周围神经系统产生了重大影响。该先证者的严重表型可归因于 PMP22 和 SOX10 基因突变的累积效应,这些突变导致脱髓鞘神经病。