Key Laboratory for Experimental Teratology of the Ministry of Education and Department of Medical Genetics, Cheeloo College of Medicine, School of Basic Medical Sciences, Shandong University, No.44 West Wenhua Road, Jinan, Shandong, 250012, People's Republic of China.
Department of Neurology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People's Republic of China.
J Neuroinflammation. 2021 Jun 28;18(1):145. doi: 10.1186/s12974-021-02193-0.
Experimental autoimmune encephalomyelitis (EAE) is an animal disease model of multiple sclerosis (MS) that involves the immune system and central nervous system (CNS). However, it is unclear how genetic predispositions promote neuroinflammation in MS and EAE. Here, we investigated how partial loss-of-function of suppressor of MEK1 (SMEK1), a regulatory subunit of protein phosphatase 4, facilitates the onset of MS and EAE.
C57BL/6 mice were immunized with myelin oligodendrocyte glycoprotein 35-55 (MOG) to establish the EAE model. Clinical signs were recorded and pathogenesis was investigated after immunization. CNS tissues were analyzed by immunostaining, quantitative polymerase chain reaction (qPCR), western blot analysis, and enzyme-linked immunosorbent assay (ELISA). Single-cell analysis was carried out in the cortices and hippocampus. Splenic and lymph node cells were evaluated with flow cytometry, qPCR, and western blot analysis.
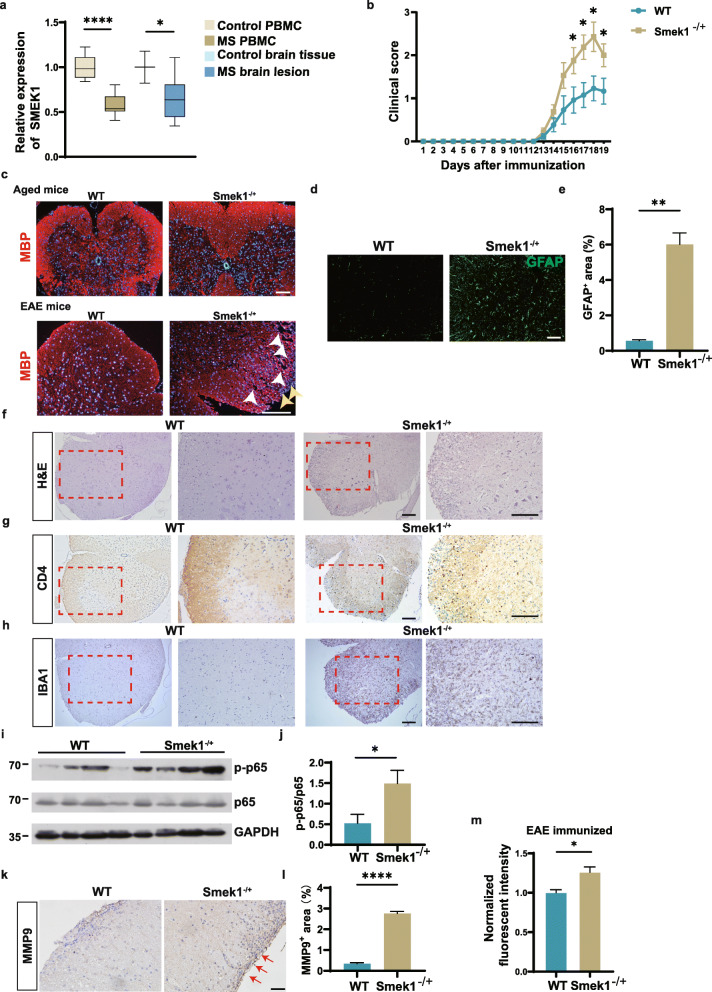
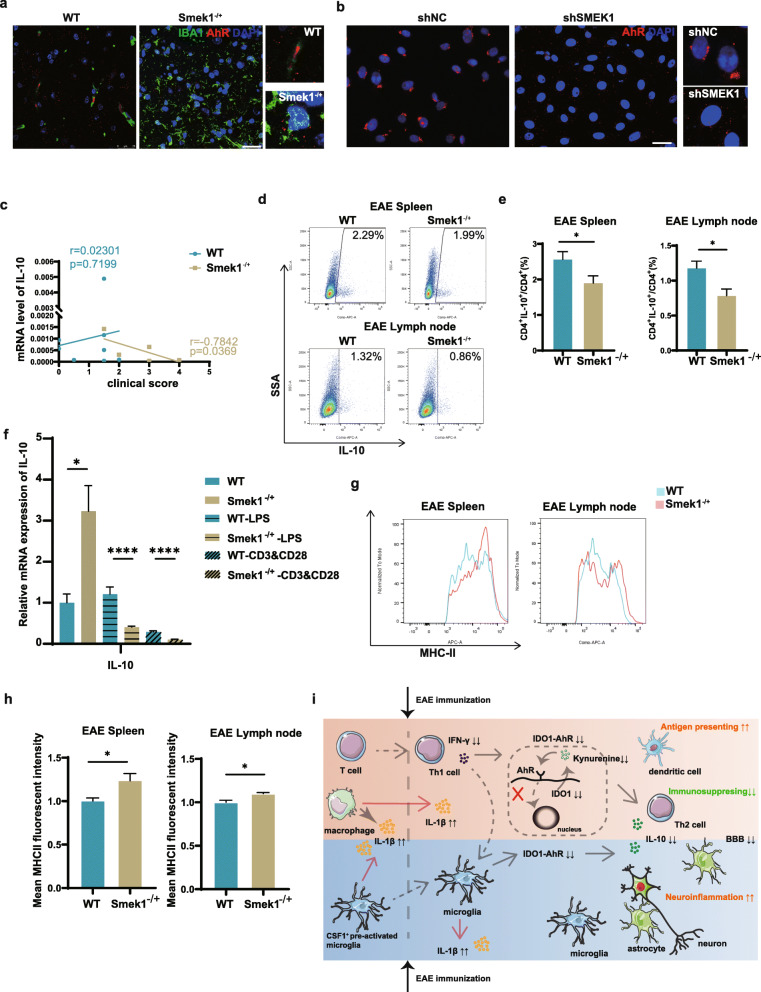
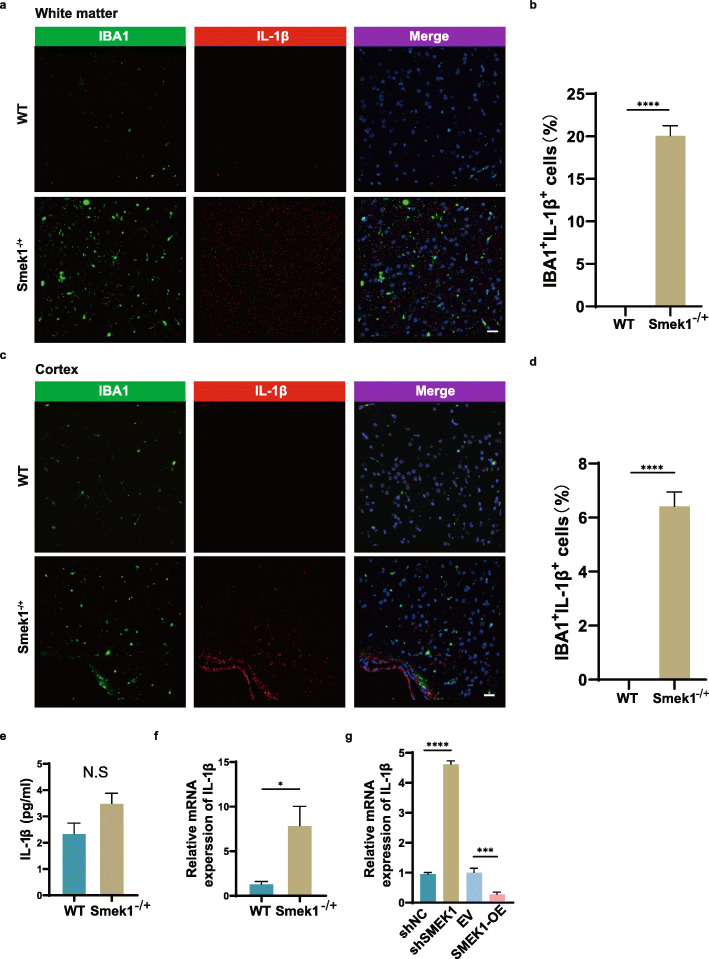
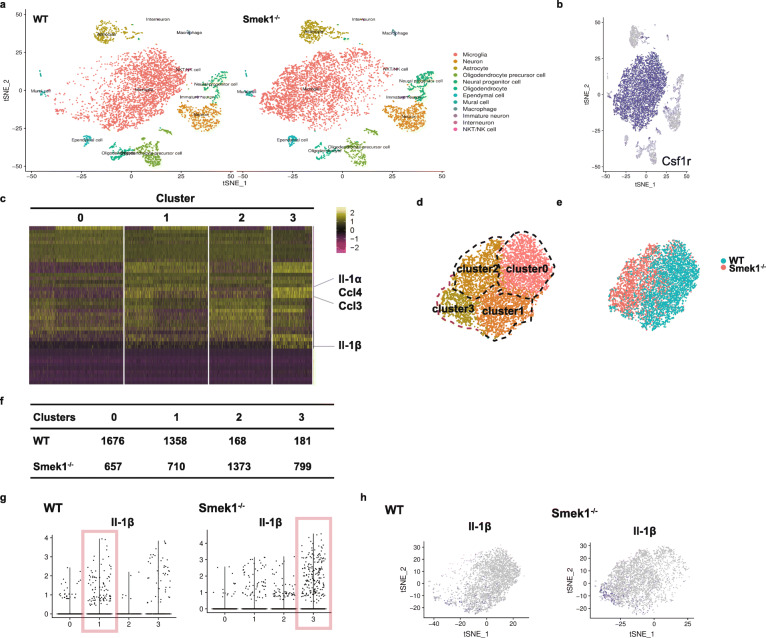
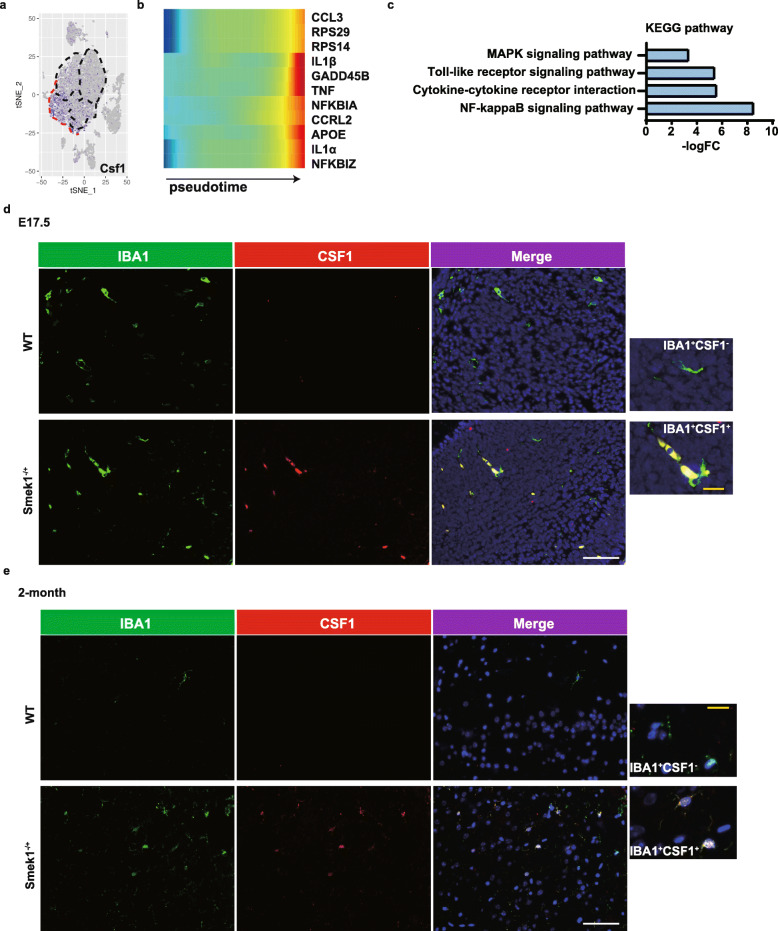
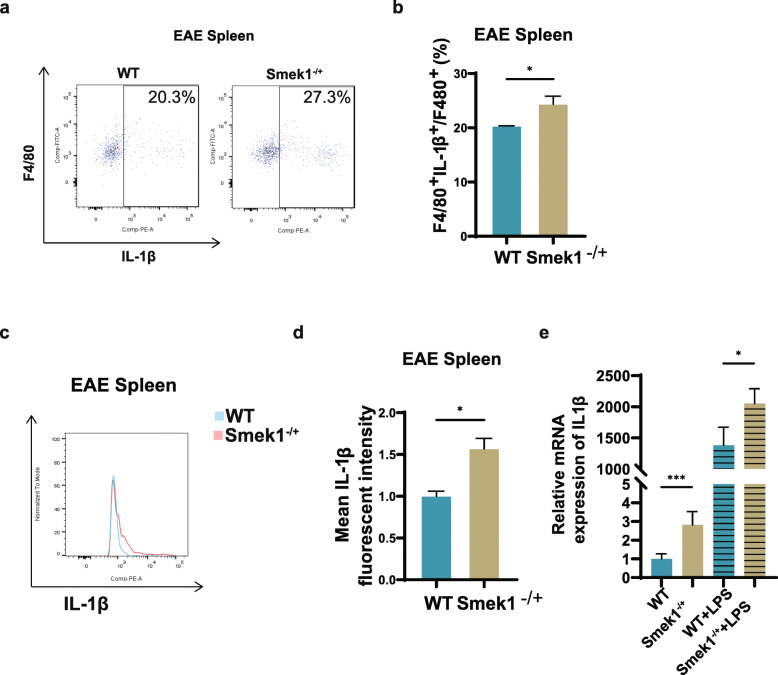
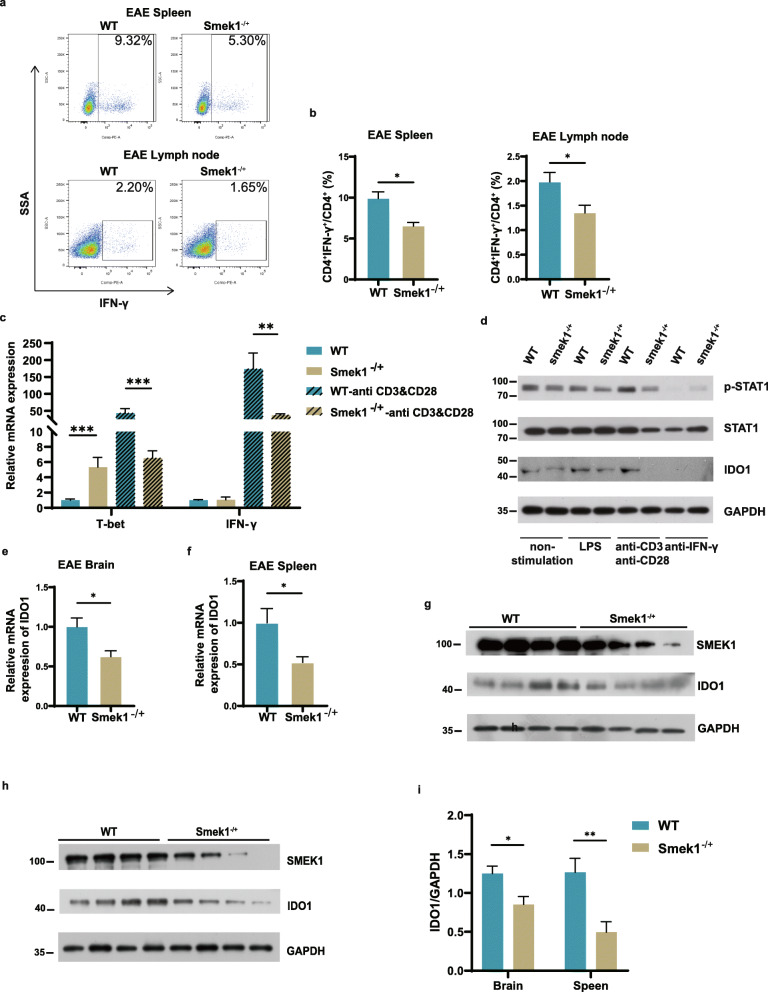
Here, we showed that partial Smek1 deficiency caused more severe symptoms in the EAE model than in controls by activating myeloid cells and that Smek1 was required for maintaining immunosuppressive function by modulating the indoleamine 2,3-dioxygenase (IDO1)-aryl hydrocarbon receptor (AhR) pathway. Single-cell sequencing and an in vitro study showed that Smek1-deficient microglia and macrophages were preactivated at steady state. After MOG immunization, microglia and macrophages underwent hyperactivation and produced increased IL-1β in Smek1 mice at the peak stage. Moreover, dysfunction of the IDO1-AhR pathway resulted from the reduction of interferon γ (IFN-γ), enhanced antigen presentation ability, and inhibition of anti-inflammatory processes in Smek1 EAE mice.
The present study suggests a protective role of Smek1 in autoimmune demyelination pathogenesis via immune suppression and inflammation regulation in both the immune system and the central nervous system. Our findings provide an instructive basis for the roles of Smek1 in EAE and broaden the understanding of the genetic factors involved in the pathogenesis of autoimmune demyelination.
实验性自身免疫性脑脊髓炎(EAE)是一种涉及免疫系统和中枢神经系统(CNS)的多发性硬化症(MS)动物疾病模型。然而,目前尚不清楚遗传易感性如何促进 MS 和 EAE 中的神经炎症。在这里,我们研究了蛋白磷酸酶 4 的调节亚基 MEK1(SMEK1)部分功能丧失如何促进 MS 和 EAE 的发病。
用髓鞘少突胶质细胞糖蛋白 35-55(MOG)免疫 C57BL/6 小鼠建立 EAE 模型。免疫后记录临床症状并进行发病机制研究。通过免疫染色、定量聚合酶链反应(qPCR)、western blot 分析和酶联免疫吸附测定(ELISA)分析中枢神经系统组织。在皮质和海马中进行单细胞分析。通过流式细胞术、qPCR 和 western blot 分析评估脾和淋巴结细胞。
在这里,我们通过激活髓样细胞表明部分 Smek1 缺陷导致 EAE 模型比对照组更严重的症状,并且 Smek1 通过调节吲哚胺 2,3-双加氧酶(IDO1)-芳香烃受体(AhR)途径来维持免疫抑制功能。单细胞测序和体外研究表明,Smek1 缺陷型小胶质细胞和巨噬细胞在稳态下预先激活。在 MOG 免疫后,Smek1 小鼠的小胶质细胞和巨噬细胞发生过度激活,并在高峰阶段产生更多的 IL-1β。此外,Smek1 EAE 小鼠中 IDO1-AhR 途径的功能障碍是由于干扰素 γ(IFN-γ)减少、抗原呈递能力增强以及抗炎过程受到抑制所致。
本研究表明,Smek1 通过在免疫系统和中枢神经系统中抑制免疫和调节炎症发挥对自身免疫性脱髓鞘发病机制的保护作用。我们的研究结果为 Smek1 在 EAE 中的作用提供了有益的依据,并拓宽了对自身免疫性脱髓鞘发病机制中遗传因素的理解。