Grupo de Xenética e Bioloxía do Desenvolvemento das Enfermidades Renais, Laboratorio de Nefroloxía (No. 11), Instituto de Investigación Sanitaria de Santiago (IDIS), Complexo Hospitalario de Santiago de Compostela (CHUS), 15706 Santiago de Compostela, Spain.
Grupo de Medicina Xenómica, Complexo Hospitalario de Santiago de Compostela (CHUS), 15706 Santiago de Compostela, Spain.
Int J Mol Sci. 2021 Jun 17;22(12):6523. doi: 10.3390/ijms22126523.
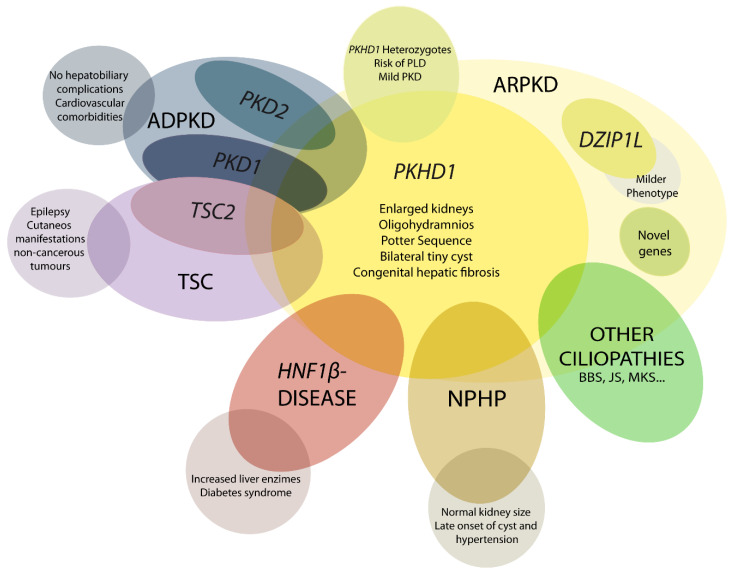
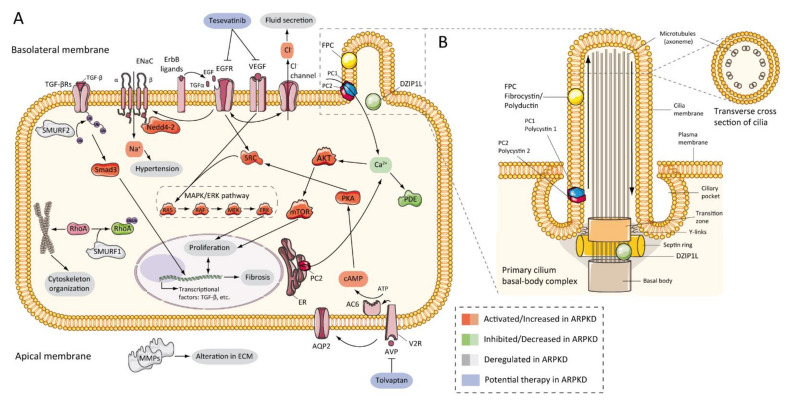
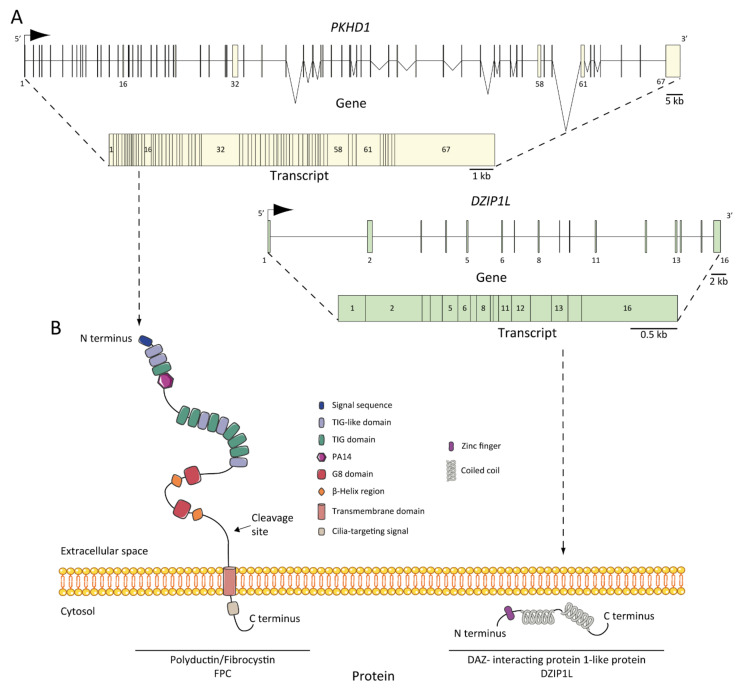
Autosomal recessive polycystic kidney disease (ARPKD) is a rare disorder and one of the most severe forms of polycystic kidney disease, leading to end-stage renal disease (ESRD) in childhood. is the gene that is responsible for the vast majority of ARPKD. However, some cases have been related to a new gene that was recently identified ( gene), as well as several ciliary genes that can mimic a ARPKD-like phenotypic spectrum. In addition, a number of molecular pathways involved in the ARPKD pathogenesis and progression were elucidated using cellular and animal models. However, the function of the ARPKD proteins and the molecular mechanism of the disease currently remain incompletely understood. Here, we review the clinics, treatment, genetics, and molecular basis of ARPKD, highlighting the most recent findings in the field.
常染色体隐性遗传性多囊肾病(ARPKD)是一种罕见疾病,也是多囊肾病中最严重的形式之一,可导致儿童期终末期肾病(ESRD)。是导致绝大多数 ARPKD 的基因。然而,一些病例与最近发现的一个新基因(基因)以及几个可以模拟 ARPKD 样表型谱的纤毛基因有关。此外,使用细胞和动物模型阐明了参与 ARPKD 发病机制和进展的许多分子途径。然而,ARPKD 蛋白的功能和疾病的分子机制目前仍不完全清楚。在这里,我们回顾了 ARPKD 的临床、治疗、遗传学和分子基础,强调了该领域的最新发现。