Pecorelli Alessandra, Cordone Valeria, Schiavone Maria Lucia, Caffarelli Carla, Cervellati Carlo, Cerbone Gaetana, Gonnelli Stefano, Hayek Joussef, Valacchi Giuseppe
Animal Science Department, Plants for Human Health Institute, North Carolina State University, Kannapolis, NC 28081, USA.
Department of Neuroscience and Rehabilitation, University of Ferrara, 44121 Ferrara, Italy.
Life (Basel). 2021 Jun 3;11(6):521. doi: 10.3390/life11060521.
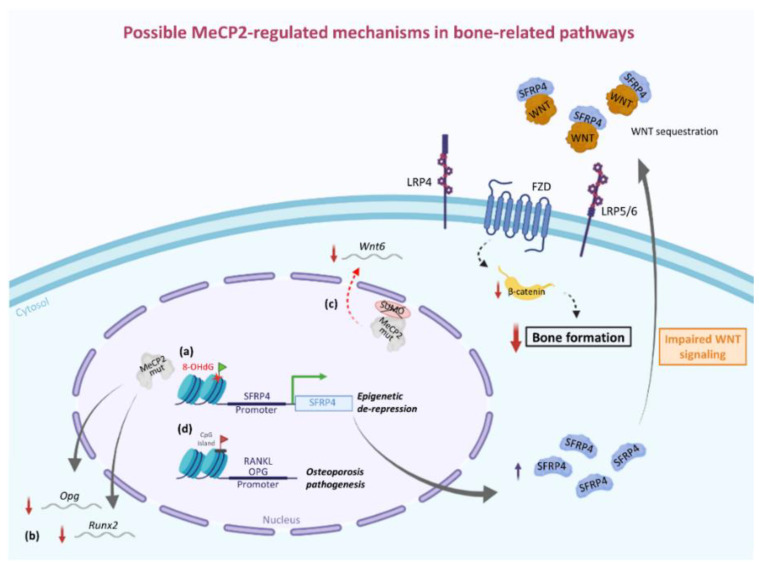
Rett syndrome (RTT) is a monogenic neurodevelopmental disorder primarily caused by mutations in X-linked gene, encoding for methyl-CpG binding protein 2 (MeCP2), a multifaceted modulator of gene expression and chromatin organization. Based on the type of mutation, RTT patients exhibit a broad spectrum of clinical phenotypes with various degrees of severity. In addition, as a complex multisystem disease, RTT shows several clinical manifestations ranging from neurological to non-neurological symptoms. The most common non-neurological comorbidities include, among others, orthopedic complications, mainly scoliosis but also early osteopenia/osteoporosis and a high frequency of fractures. A characteristic low bone mineral density dependent on a slow rate of bone formation due to dysfunctional osteoblast activity rather than an increase in bone resorption is at the root of these complications. Evidence from human and animal studies supports the idea that mutation could be associated with altered epigenetic regulation of bone-related factors and signaling pathways, including SFRP4/WNT/β-catenin axis and RANKL/RANK/OPG system. More research is needed to better understand the role of MeCP2 in bone homeostasis. Indeed, uncovering the molecular mechanisms underlying RTT bone problems could reveal new potential pharmacological targets for the treatment of these complications that adversely affect the quality of life of RTT patients for whom the only therapeutic approaches currently available include bisphosphonates, dietary supplements, and physical activity.
瑞特综合征(RTT)是一种单基因神经发育障碍疾病,主要由X连锁基因突变引起,该基因编码甲基CpG结合蛋白2(MeCP2),这是一种对基因表达和染色质组织具有多方面调节作用的蛋白。根据突变类型,RTT患者表现出广泛的临床表型,严重程度各不相同。此外,作为一种复杂的多系统疾病,RTT表现出从神经症状到非神经症状的多种临床表现。最常见的非神经合并症包括骨科并发症,主要是脊柱侧弯,还有早期骨质减少/骨质疏松以及高骨折发生率。这些并发症的根源在于骨密度低,这取决于成骨细胞功能障碍导致的骨形成速率缓慢,而非骨吸收增加。来自人类和动物研究的证据支持这样一种观点,即突变可能与骨相关因子和信号通路(包括SFRP4/WNT/β-连环蛋白轴和RANKL/RANK/OPG系统)的表观遗传调控改变有关。需要更多研究来更好地理解MeCP2在骨稳态中的作用。事实上,揭示RTT骨问题背后的分子机制可能会揭示治疗这些并发症的新潜在药理学靶点,这些并发症对RTT患者的生活质量产生不利影响,而目前唯一可用的治疗方法包括双膦酸盐、膳食补充剂和体育活动。