Štekláč Marek, Zajaček Dávid, Bučinský Lukáš
Institute of Physical Chemistry and Chemical Physics, Faculty of Chemical and Food Technology, Slovak Technical University, Bratislava SK-81237, Slovakia.
J Mol Struct. 2021 Dec 5;1245:130968. doi: 10.1016/j.molstruc.2021.130968. Epub 2021 Jun 28.
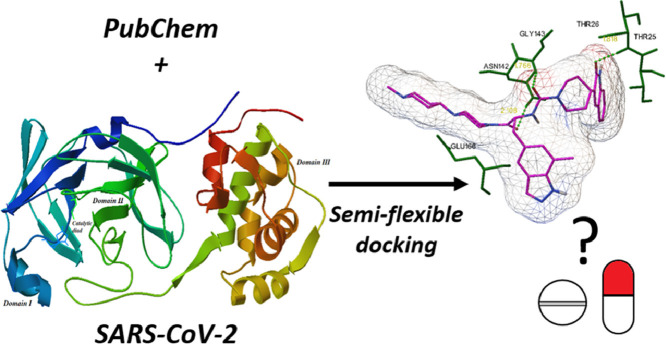
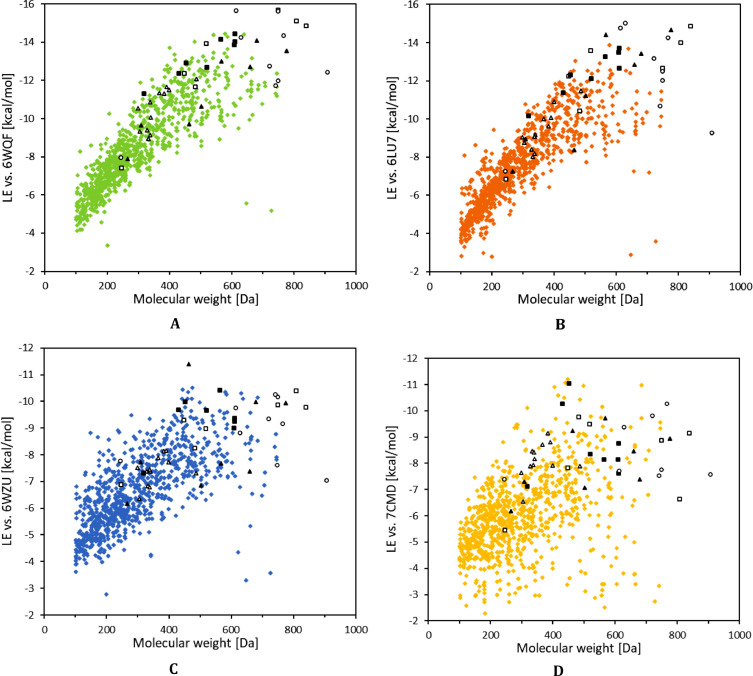
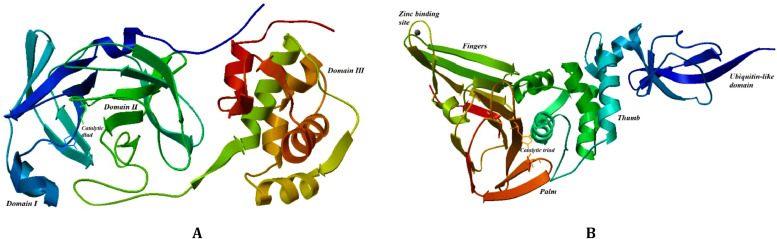
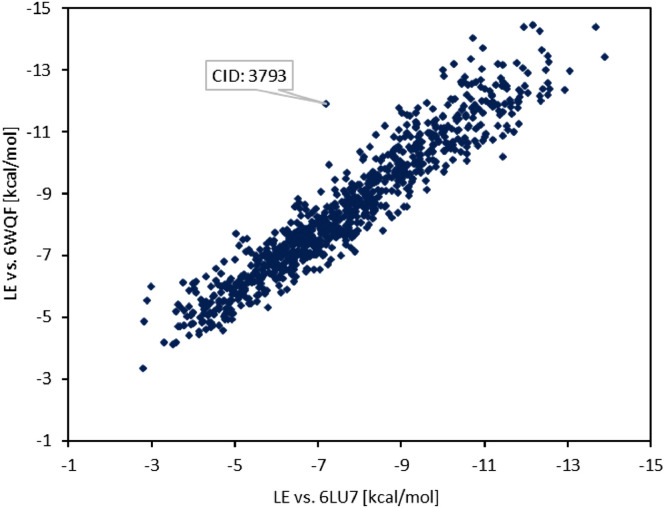
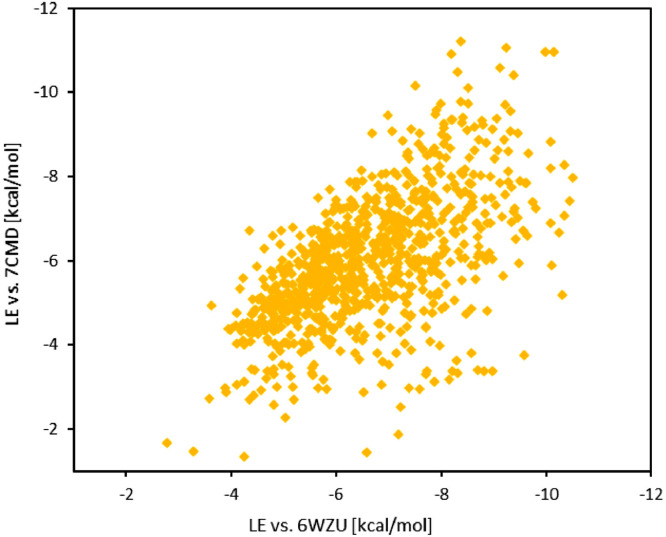
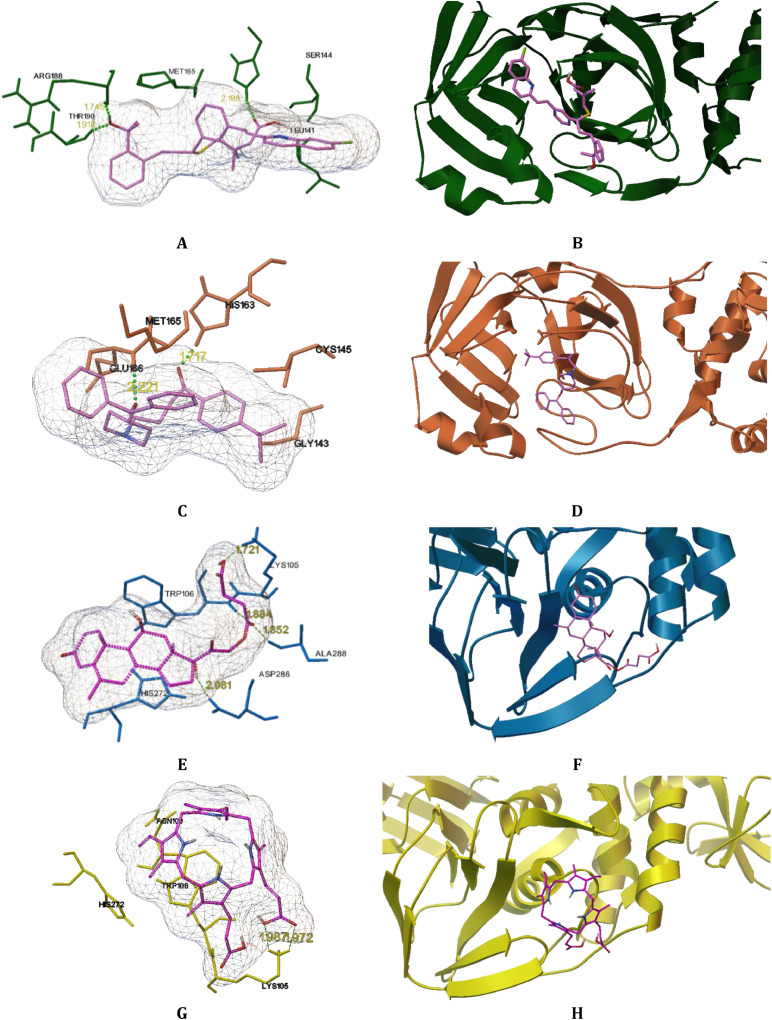
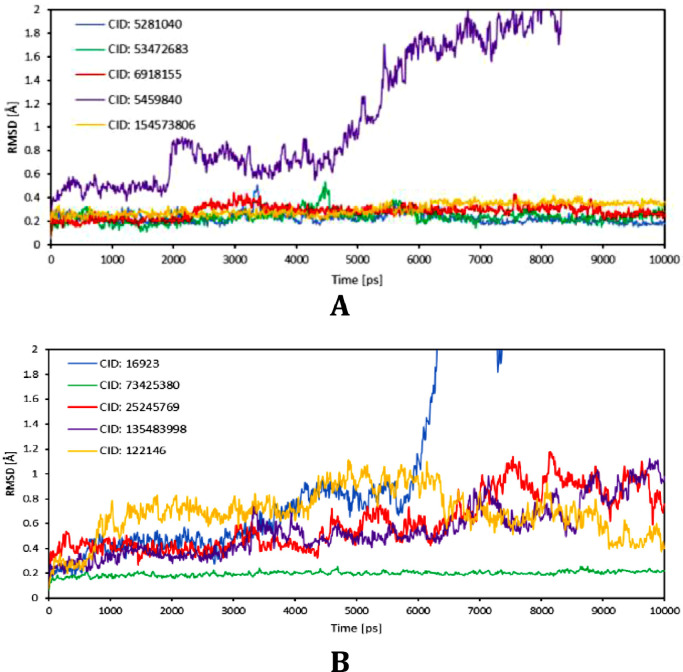
The spread of a novel coronavirus SARS-CoV-2 and a resulting COVID19 disease in late 2019 has transformed into a worldwide pandemic and has effectively brought the world to a halt. Proteases 3CL and PL, responsible for proteolysis of new virions, represent vital inhibition targets for the COVID19 treatment. Herein, we report an docking study of more than 860 COVID19-related compounds from the PubChem database. Molecular dynamic simulations were carried out to validate the conformation stability of compound-ligand complexes with best docking scores. The MM-PBSA approach was employed to calculate binding free energies. The comparison with ca. 50 previously reported potential SARS-CoV-2's proteases inhibitors show a number of new compounds with excellent binding affinities. Anti-inflammatory drugs Montelukast, Ebastine and Solumedrol, the anti-migraine drug Vazegepant or the anti-MRSA pro-drug TAK-599, among many others, all show remarkable affinities to 3CL and with known side effects present candidates for immediate clinical trials. This study reports thorough docking scores summary of COVID19-related compounds found in the PubChem database and illustrates the asset of computational screening methods in search for possible drug-like candidates. Several yet-untested compounds show affinities on par with reported inhibitors and warrant further attention. Furthermore, the submitted work provides readers with ADME data, ZINC and PubChem IDs, as well as docking scores of all studied compounds for further comparisons.
2019年末,新型冠状病毒SARS-CoV-2的传播以及由此引发的COVID-19疾病已演变成一场全球大流行,并实际上使世界陷入停滞。负责新病毒粒子蛋白水解的蛋白酶3CL和PL是COVID-19治疗的重要抑制靶点。在此,我们报告了一项对来自PubChem数据库的860多种与COVID-19相关化合物的对接研究。进行了分子动力学模拟以验证具有最佳对接分数的化合物-配体复合物的构象稳定性。采用MM-PBSA方法计算结合自由能。与约50种先前报道的潜在SARS-CoV-2蛋白酶抑制剂的比较表明,有许多具有优异结合亲和力的新化合物。抗炎药孟鲁司特、依巴斯汀和甲泼尼龙、抗偏头痛药伐地昔帕、抗耐甲氧西林金黄色葡萄球菌前药TAK-599等,都对3CL表现出显著的亲和力,且由于存在已知的副作用,成为立即进行临床试验的候选药物。本研究报告了在PubChem数据库中发现的与COVID-19相关化合物的详细对接分数总结,并说明了计算筛选方法在寻找可能的类药物候选物方面的作用。几种尚未测试的化合物表现出与已报道抑制剂相当的亲和力,值得进一步关注。此外,提交的工作为读者提供了所有研究化合物的ADME数据、ZINC和PubChem ID以及对接分数,以供进一步比较。