Division of Cardiothoracic Surgery, Rhode Island Hospital, Alpert Medical School of Brown University, Providence, RI.
Department of Medicine, Vascular Research Laboratory, Providence VA Medical Center, Alpert Medical School of Brown University, Providence, RI.
J Thorac Cardiovasc Surg. 2022 Nov;164(5):e207-e226. doi: 10.1016/j.jtcvs.2021.06.029. Epub 2021 Jun 26.
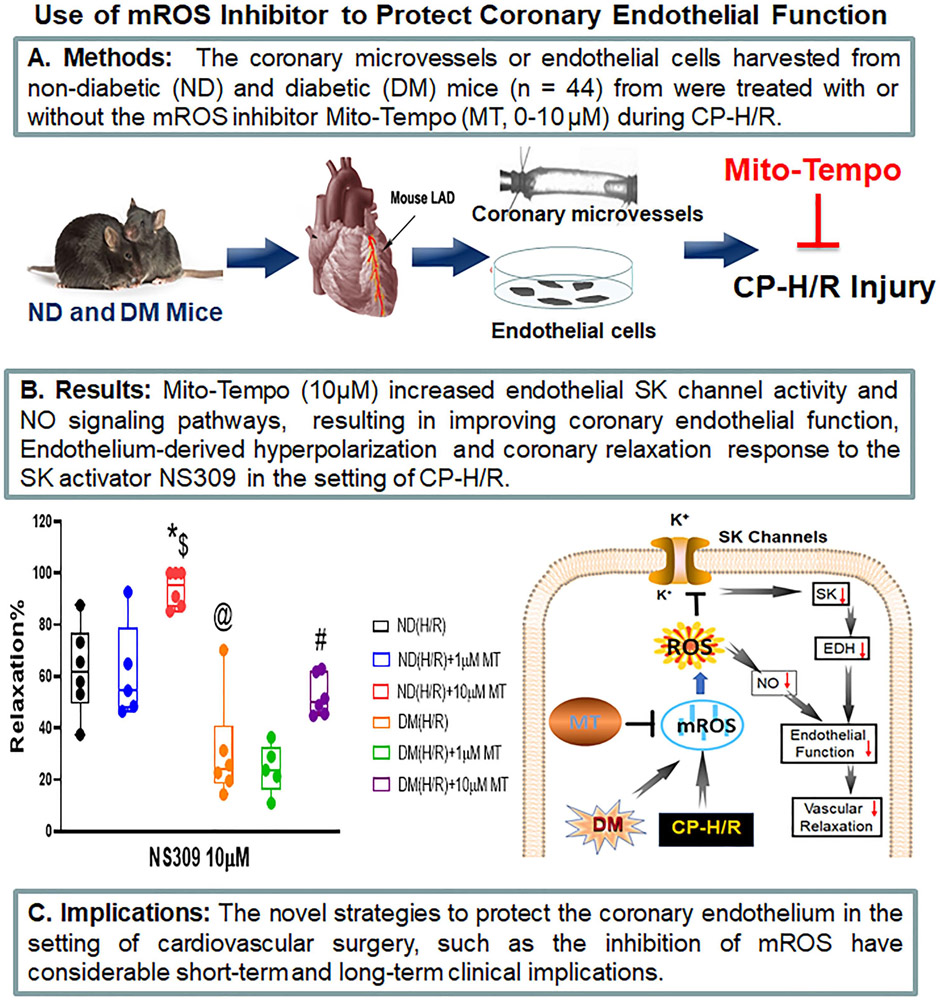
Cardioplegic ischemia-reperfusion and diabetes mellitus are correlated with coronary endothelial dysfunction and inactivation of small conductance calcium-activated potassium channels. Increased reactive oxidative species, such as mitochondrial reactive oxidative species, may contribute to oxidative injury. Thus, we hypothesized that inhibition of mitochondrial reactive oxidative species may protect coronary small conductance calcium-activated potassium channels and endothelial function against cardioplegic ischemia-reperfusion-induced injury.
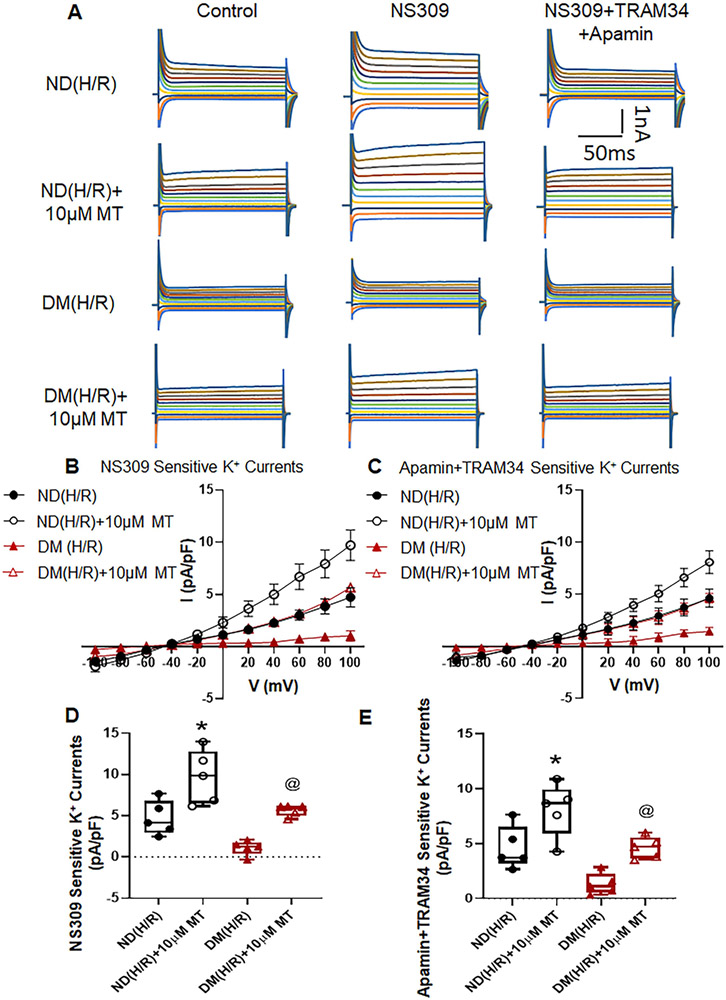
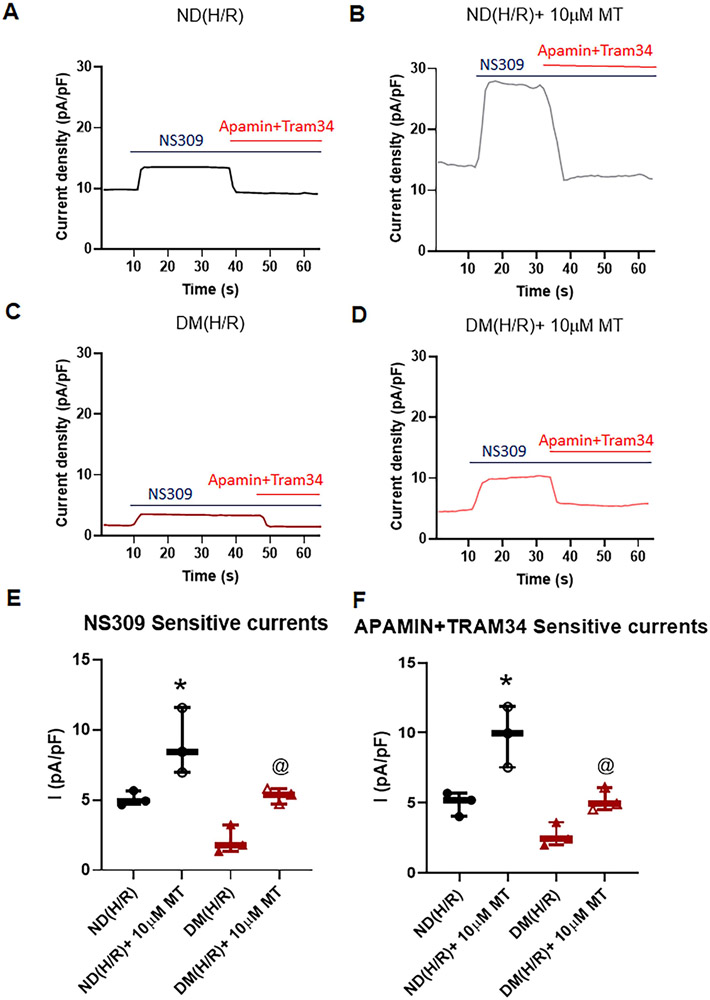
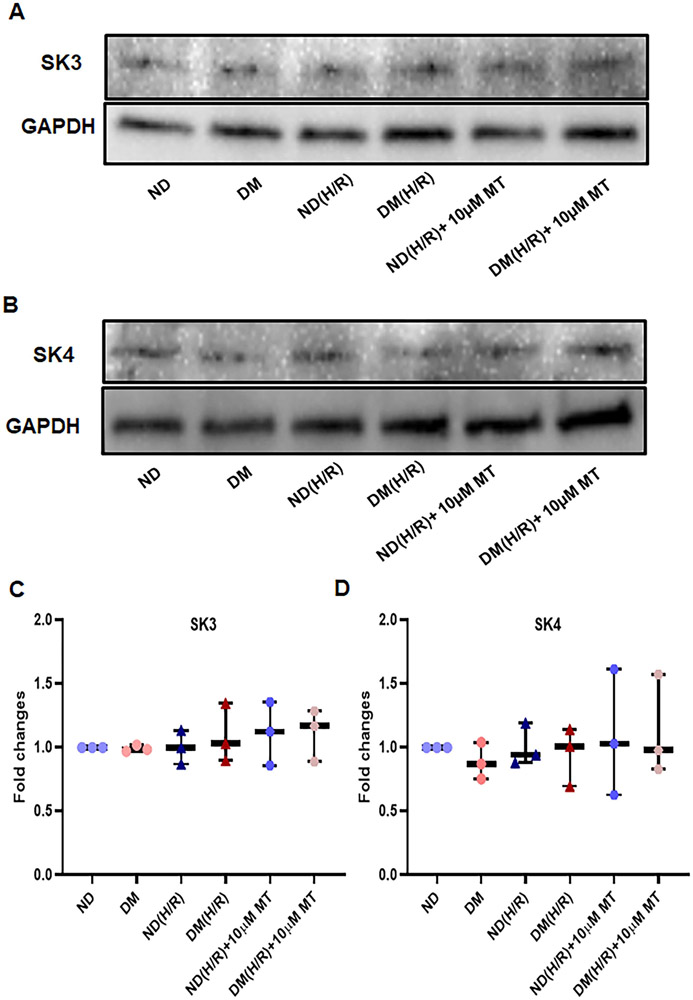
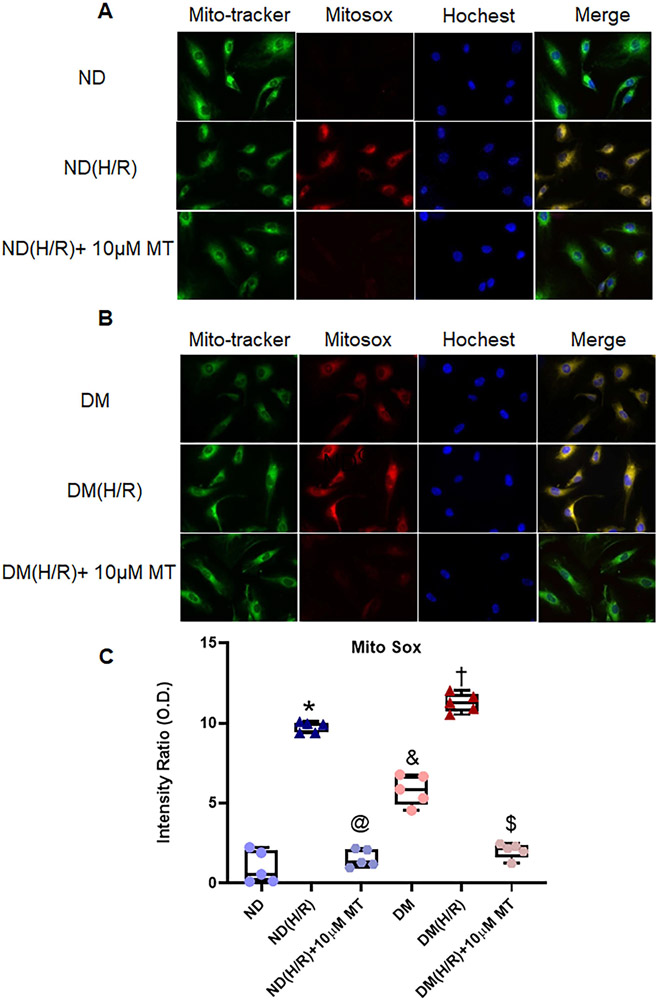
Small coronary arteries and endothelial cells from the hearts of mice with and without diabetes mellitus were isolated and examined by using a cardioplegic hypoxia and reoxygenation model to determine whether the mitochondria-targeted antioxidant Mito-Tempo could protect against coronary endothelial and small conductance calcium-activated potassium channel dysfunction. The microvessels or mouse heart endothelial cells were treated with or without Mito-Tempo (0-10 μM) 5 minutes before and during cardioplegic hypoxia and reoxygenation. Microvascular function was assessed in vitro by vessel myography. K currents of mouse heart endothelial cells were measured by whole-cell patch clamp. The levels of intracellular cytosolic free calcium (Ca) concentration, mitochondrial reactive oxidative species, and small conductance calcium-activated potassium protein expression of mouse heart endothelial cells were measured by Rhod-2 fluorescence staining, MitoSox, and Western blotting, respectively.
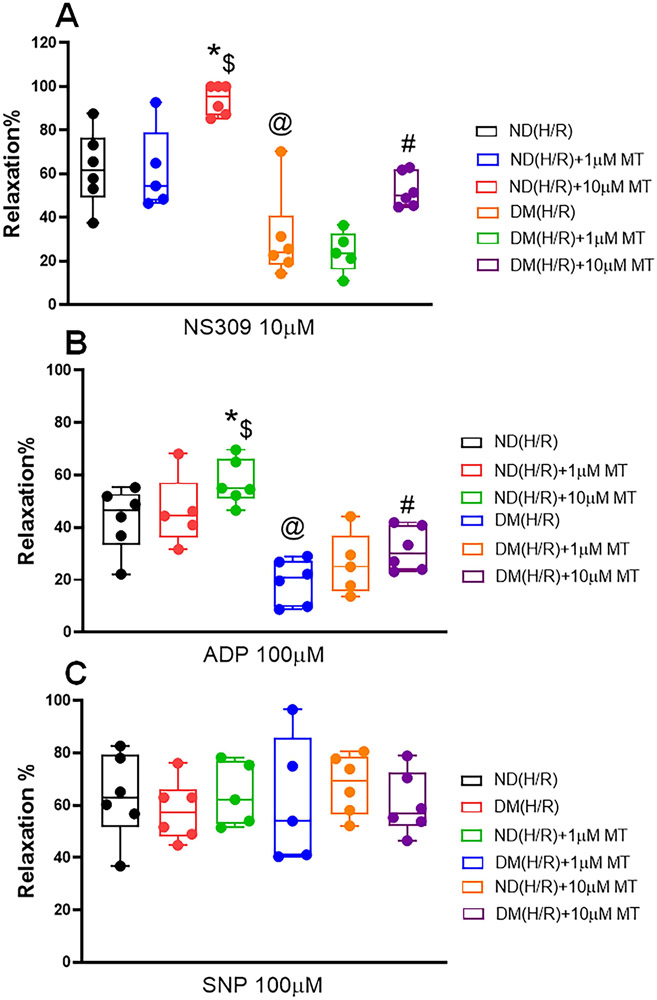
Cardioplegic hypoxia and reoxygenation significantly attenuated endothelial small conductance calcium-activated potassium channel activity, caused calcium overload, and increased mitochondrial reactive oxidative species of mouse heart endothelial cells in both the nondiabetic and diabetes mellitus groups. In addition, treating mouse heart endothelial cells with Mito-Tempo (10 μM) reduced cardioplegic hypoxia and reoxygenation-induced Ca and mitochondrial reactive oxidative species overload in both the nondiabetic and diabetes mellitus groups, respectively (P < .05). Treatment with Mito-Tempo (10 μM) significantly enhanced coronary relaxation responses to adenosine 5'-diphosphate and NS309 (P < .05), and endothelial small conductance calcium-activated potassium channel currents in both the nondiabetic and diabetes mellitus groups (P < .05).
Administration of Mito-Tempo improves endothelial function and small conductance calcium-activated potassium channel activity, which may contribute to its enhancement of endothelium-dependent vasorelaxation after cardioplegic hypoxia and reoxygenation.
心脏停搏性缺血再灌注和糖尿病与冠状动脉内皮功能障碍和小电导钙激活钾通道失活有关。活性氧物质(如线粒体活性氧物质)的增加可能导致氧化损伤。因此,我们假设抑制线粒体活性氧物质可能会保护冠状动脉小电导钙激活钾通道和内皮功能免受心脏停搏性缺血再灌注引起的损伤。
使用心脏停搏性缺氧再复氧模型分离并检测糖尿病和非糖尿病小鼠的小冠状动脉和内皮细胞,以确定线粒体靶向抗氧化剂 Mito-Tempo 是否可以保护冠状动脉内皮和小电导钙激活钾通道功能障碍。在心脏停搏性缺氧和再复氧之前和期间,用或不用 Mito-Tempo(0-10 μM)处理微血管或小鼠心脏内皮细胞 5 分钟。通过血管肌描记术在体外评估微血管功能。通过全细胞膜片钳测量小鼠心脏内皮细胞的 K 电流。通过 Rhod-2 荧光染色、MitoSox 和 Western blot 分别测量小鼠心脏内皮细胞的细胞内细胞质游离钙(Ca)浓度、线粒体活性氧物质和小电导钙激活钾蛋白表达水平。
心脏停搏性缺氧和再复氧显著减弱了内皮小电导钙激活钾通道活性,导致钙超载,并增加了非糖尿病和糖尿病小鼠心脏内皮细胞的线粒体活性氧物质。此外,用 Mito-Tempo(10 μM)处理小鼠心脏内皮细胞可分别减少非糖尿病和糖尿病小鼠心脏内皮细胞的心脏停搏性缺氧和再复氧引起的 Ca 和线粒体活性氧物质过载(P <.05)。用 Mito-Tempo(10 μM)处理可显著增强非糖尿病和糖尿病小鼠心脏内皮细胞对腺苷 5'-二磷酸和 NS309 的冠状血管舒张反应(P <.05)和内皮小电导钙激活钾通道电流(P <.05)。
给予 Mito-Tempo 可改善内皮功能和小电导钙激活钾通道活性,这可能有助于其增强心脏停搏性缺氧和再复氧后的内皮依赖性血管舒张作用。