Falsified Medicines and Medical Devices Department, National Medicines Institute, Chełmska 30/34, 00-725 Warsaw, Poland.
Department of Synthetic Drugs, National Medicines Institute, Chełmska 30/34, 00-725 Warsaw, Poland.
Int J Mol Sci. 2021 Jul 1;22(13):7139. doi: 10.3390/ijms22137139.
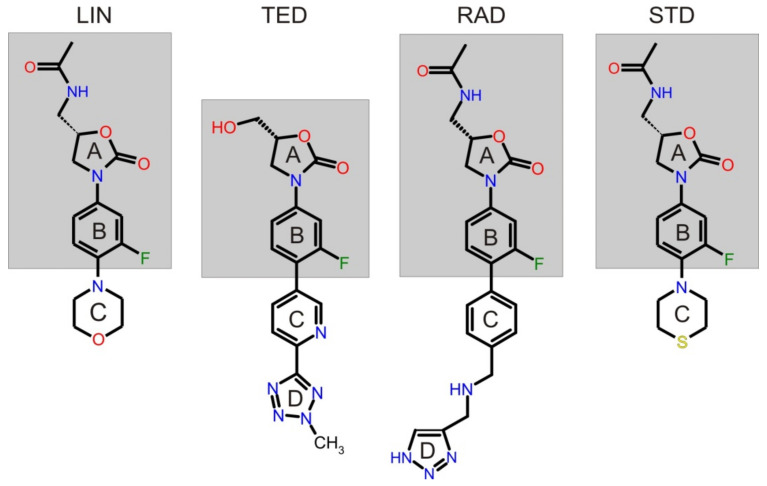
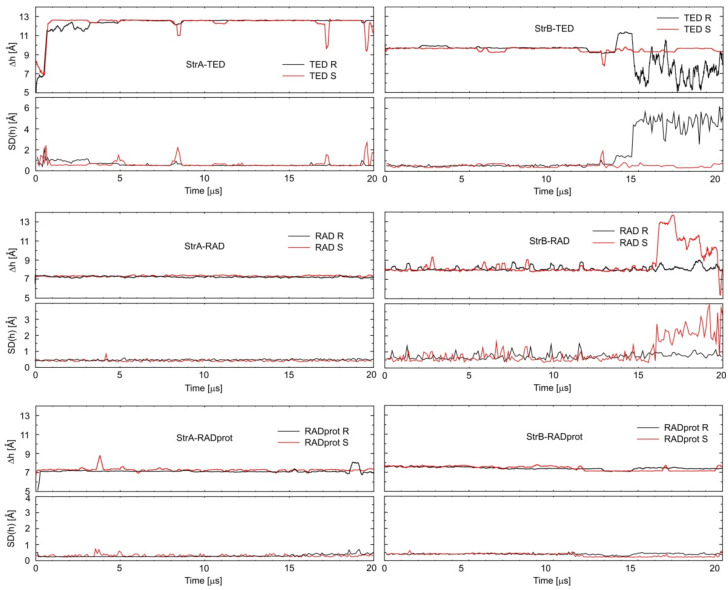
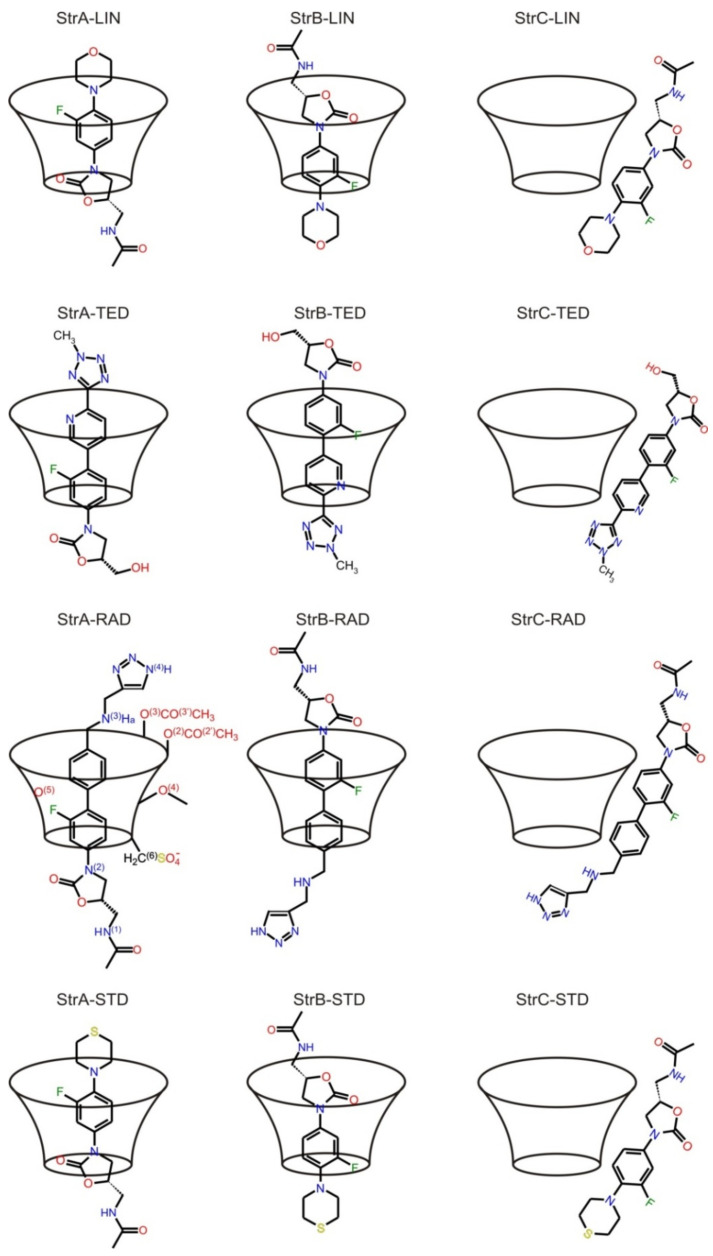
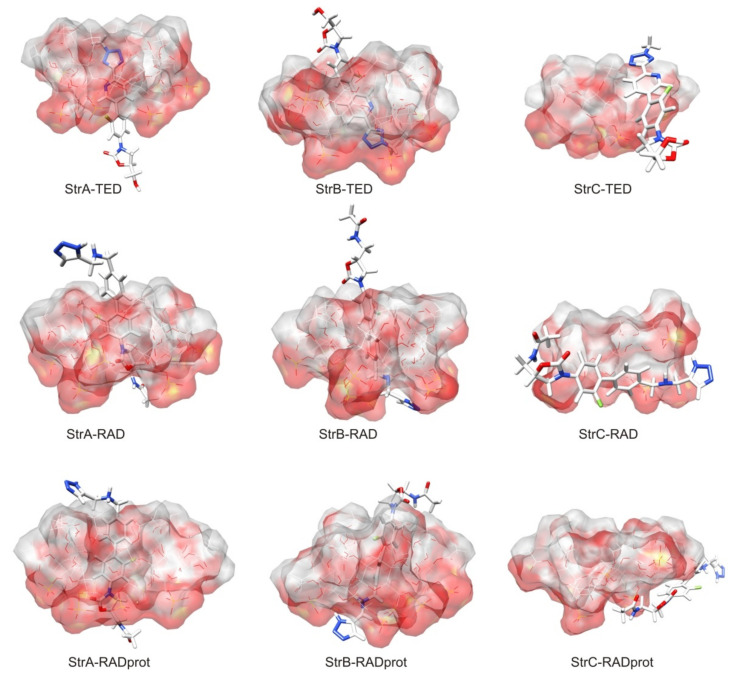
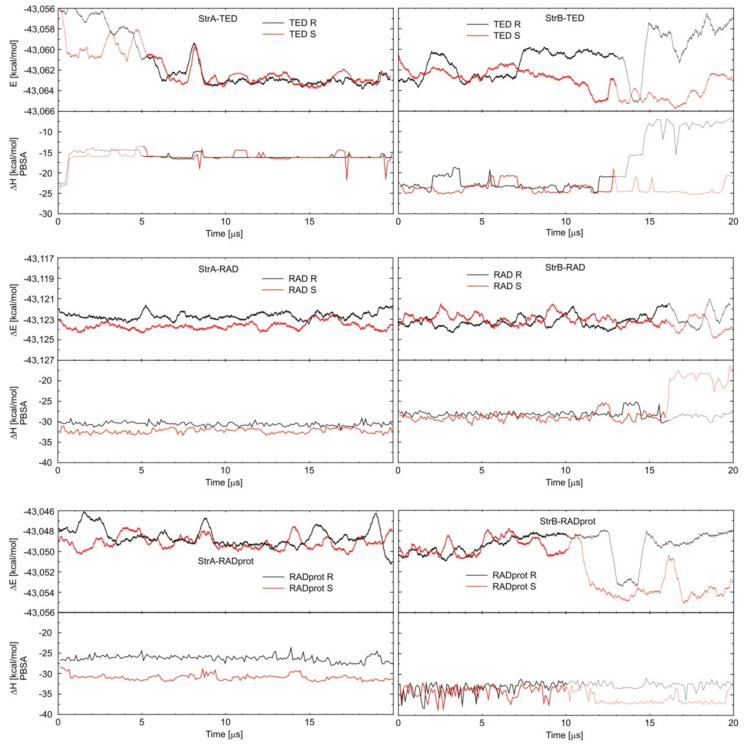
Molecular modeling (MM) results for tedizolid and radezolid with heptakis-(2,3-diacetyl-6-sulfo)-β-cyclodextrin (HDAS-β-CD) are presented and compared with the results previously obtained for linezolid and sutezolid. The mechanism of interaction of chiral oxazolidinone ligands belonging to a new class of antibacterial agents, such as linezolid, tedizolid, radezolid, and sutezolid, with HDAS-β-CD based on capillary electrokinetic chromatography (cEKC), nuclear magnetic resonance (NMR) spectroscopy, and MM methods was described. Principles of chiral separation of oxazolidinone analogues using charged single isomer derivatives of cyclodextrin by the cEKC method were presented, including the selection of the optimal chiral selector and separation conditions, complex stoichiometry, and binding constants, which provided a comprehensive basis for MM studies. In turn, NMR provided, where possible, direct information on the geometry of the inclusion complexes and also provided the necessary structural information to validate the MM calculations. Consequently, MM contributed to the understanding of the structure of diastereomeric complexes, the thermodynamics of complexation, and the visualization of their structures. The most probable mean geometries of the studied supramolecular complexes and their dynamics (geometry changes over time) were determined by molecular dynamics methods. Oxazolidinone ligands have been shown to complex mainly the inner part of cyclodextrin, while the external binding is less privileged, which is consistent with the conclusions of the NMR studies. Enthalpy values of binding of complexes were calculated using long-term molecular dynamics in explicit water as well as using molecular mechanics, the Poisson-Boltzmann or generalized Born, and surface area continuum solvation (MM/PBSA and MM/GBSA) methods. Computational methods predicted the effect of changes in pH and composition of the solution on the strength and complexation process, and it adapted the conditions selected as optimal during the cEKC study. By changing the dielectric constant in the MM/PBSA and MM/GBSA calculations, the effect of changing the solution to methanol/acetonitrile was investigated. A fairly successful attempt was made to predict the chiral separation of the oxazolidinones using the modified cyclodextrin by computational methods.
呈现了替加环素和瑞德昔布与七(2,3-二乙酰基-6-磺酸基)-β-环糊精(HDAS-β-CD)的分子建模(MM)结果,并与先前获得的利奈唑胺和苏替唑胺的结果进行了比较。描述了属于新型抗菌剂的手性恶唑烷酮配体(如利奈唑胺、替加环素、瑞德昔布和苏替唑胺)与基于毛细管电泳(CEKC)、核磁共振(NMR)光谱和 MM 方法的 HDAS-β-CD 的相互作用机制。提出了利用环糊精的带电单异构体衍生物通过 CEKC 方法对手性分离恶唑烷酮类似物的原理,包括选择最佳手性选择器和分离条件、络合化学计量和结合常数,为 MM 研究提供了全面的基础。反过来,NMR 提供了包含配合物的几何形状的直接信息,并且还提供了验证 MM 计算所需的结构信息。因此,MM 有助于理解非对映体配合物的结构、络合的热力学以及它们的结构可视化。通过分子动力学方法确定了研究超分子配合物的最可能的平均几何形状及其动力学(随时间的几何形状变化)。恶唑烷酮配体主要与环糊精的内部结合,而外部结合则不太有利,这与 NMR 研究的结论一致。使用长期分子动力学在显式水中以及使用分子力学、泊松-玻尔兹曼或广义 Born 和表面积连续体溶剂化(MM/PBSA 和 MM/GBSA)方法计算了络合物结合的焓值。计算方法预测了改变 pH 值和溶液组成对强度和络合过程的影响,并适应了在 CEKC 研究中选择的最佳条件。通过在 MM/PBSA 和 MM/GBSA 计算中改变介电常数,研究了改变溶液为甲醇/乙腈的效果。通过计算方法对手性分离恶唑烷酮进行了相当成功的尝试。