Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
J Am Chem Soc. 2021 Aug 4;143(30):11337-11344. doi: 10.1021/jacs.1c06287. Epub 2021 Jul 21.
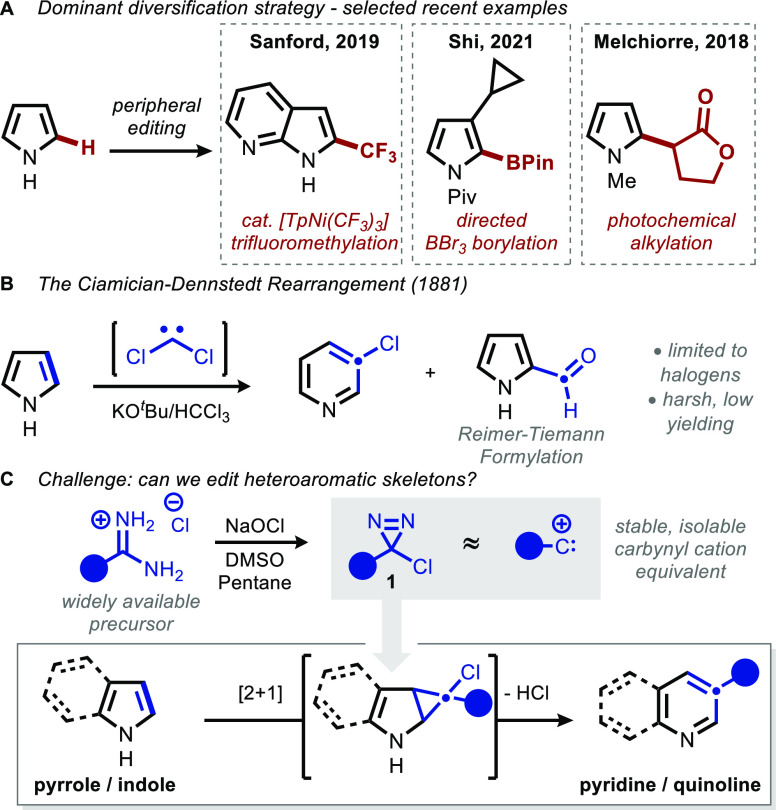
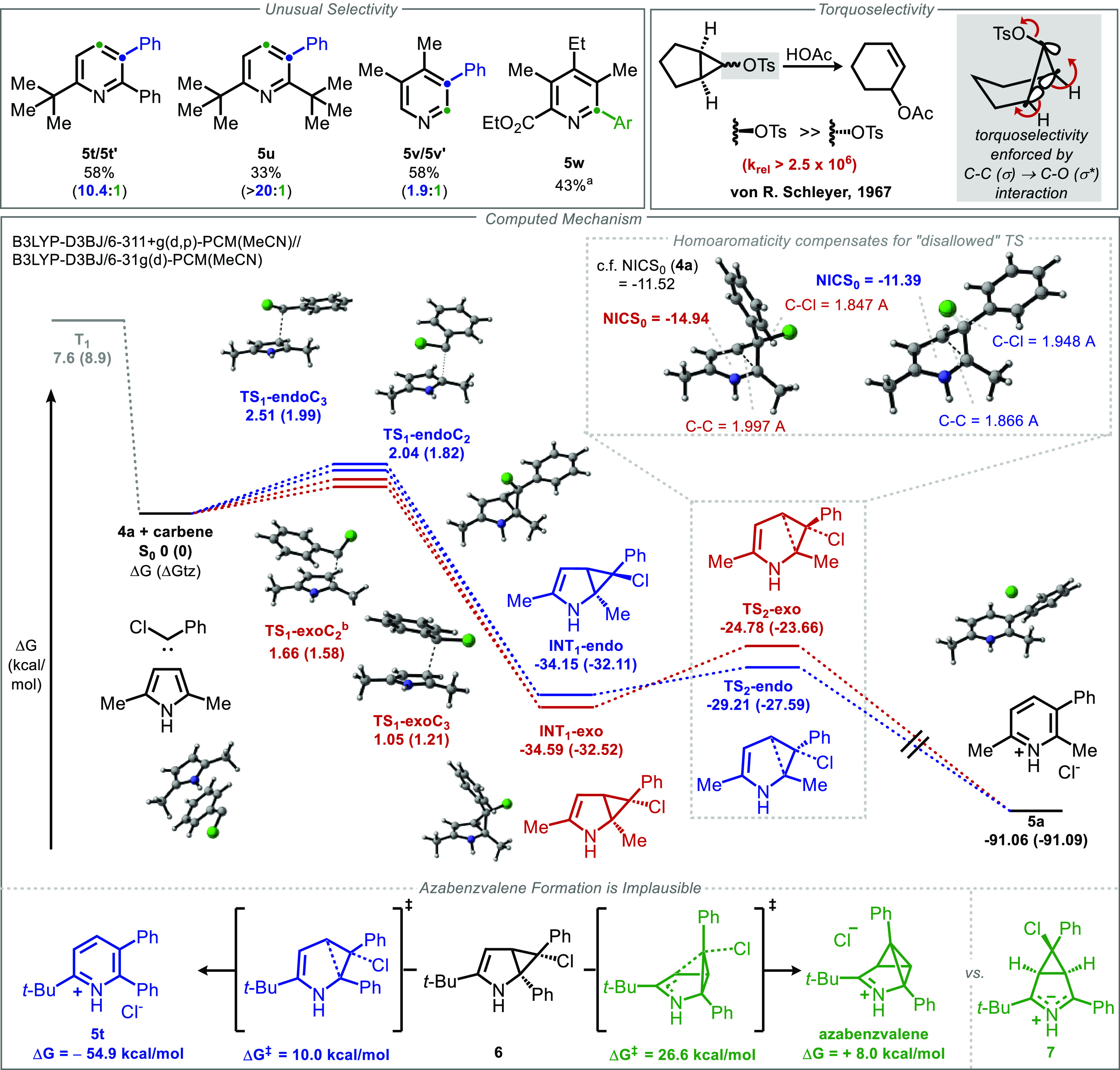
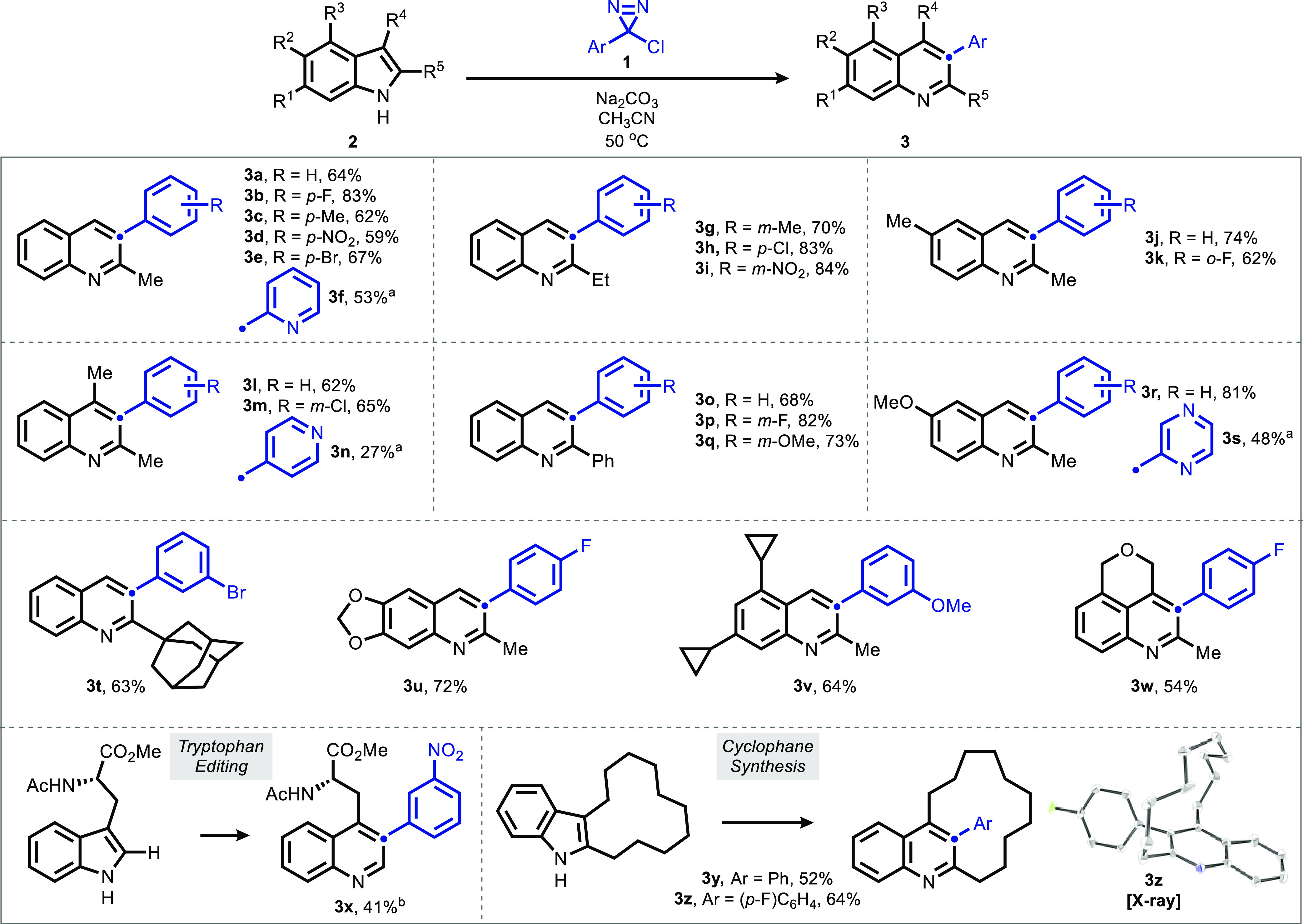
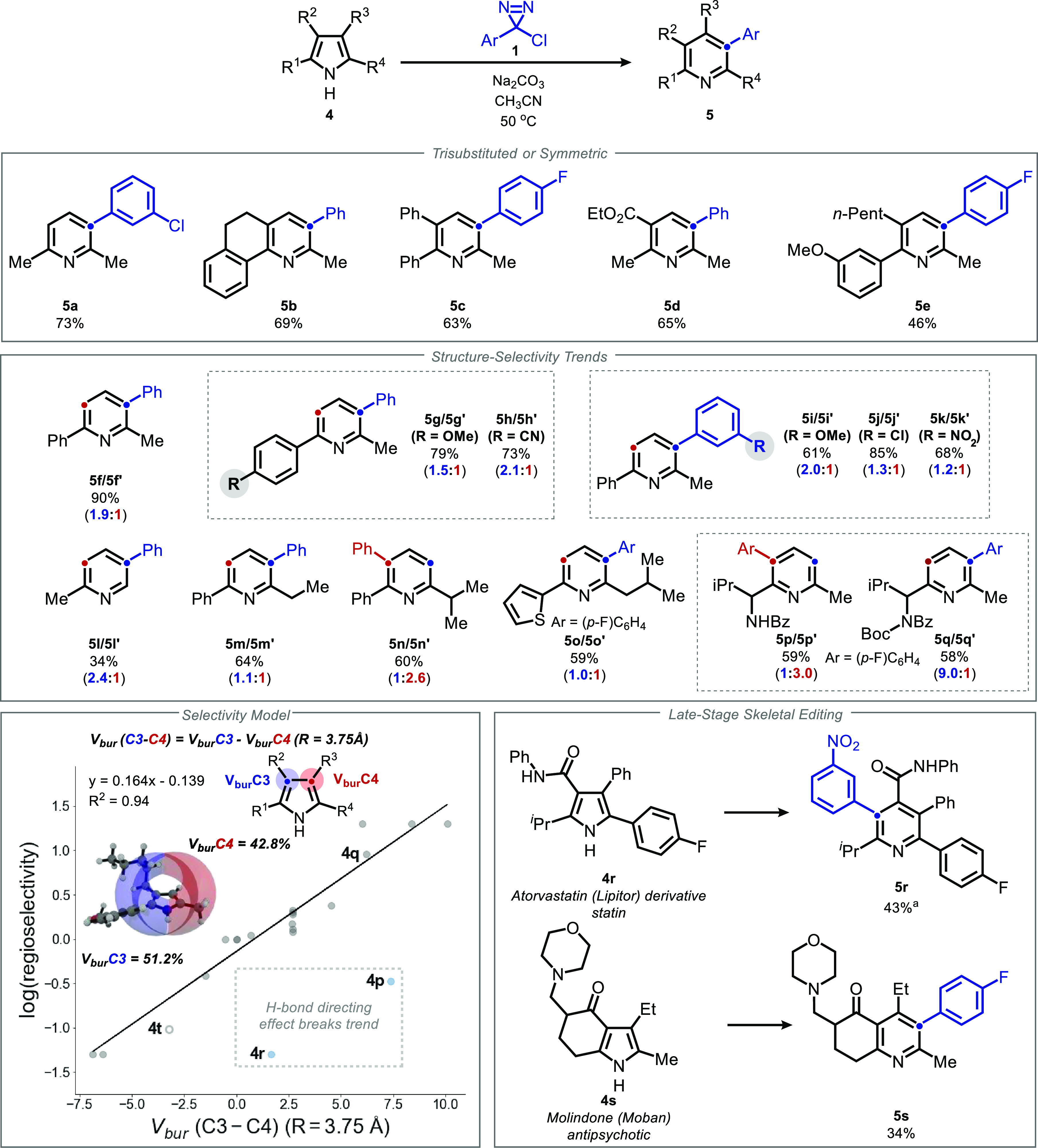
Herein, we report a reaction that selectively generates 3-arylpyridine and quinoline motifs by inserting aryl carbynyl cation equivalents into pyrrole and indole cores, respectively. By employing α-chlorodiazirines as thermal precursors to the corresponding chlorocarbenes, the traditional haloform-based protocol central to the parent Ciamician-Dennstedt rearrangement can be modified to directly afford 3-(hetero)arylpyridines and quinolines. Chlorodiazirines are conveniently prepared in a single step by oxidation of commercially available amidinium salts. Selectivity as a function of pyrrole substitution pattern was examined, and a predictive model based on steric effects is put forward, with DFT calculations supporting a selectivity-determining cyclopropanation step. Computations surprisingly indicate that the stereochemistry of cyclopropanation is of little consequence to the subsequent electrocyclic ring opening that forges the pyridine core, due to a compensatory homoaromatic stabilization that counterbalances orbital-controlled torquoselectivity effects. The utility of this skeletal transform is further demonstrated through the preparation of quinolinophanes and the skeletal editing of pharmaceutically relevant pyrroles.
在这里,我们报告了一种反应,通过将芳基卡宾阳离子等价物分别插入吡咯和吲哚核中,选择性地生成 3-芳基吡啶和喹啉基序。通过使用α-氯代二氮杂环丁烷作为相应氯卡宾的热前体,传统的基于卤仿的方案可以被修改为直接得到 3-(杂)芳基吡啶和喹啉。氯代二氮杂环丁烷可以通过商业上可获得的脒盐的氧化方便地一步制备。我们研究了吡咯取代模式的选择性,并提出了一个基于空间效应的预测模型,DFT 计算支持了选择性决定的环丙烷化步骤。令人惊讶的是,计算表明,环丙烷化的立体化学对于随后形成吡啶核的电环化开环几乎没有影响,因为补偿性的同芳香稳定化抵消了轨道控制的扭转选择性效应。通过制备喹啉并和具有药物相关吡咯的骨架编辑,进一步证明了这种骨架转化的实用性。