Judicate George P, Barabona Godfrey, Kamori Doreen, Mahiti Macdonald, Tan Toong Seng, Ozono Seiya, Mgunya Amina Shaban, Kuwata Takeo, Matsushita Shuzo, Sunguya Bruno, Lyamuya Eligius, Tokunaga Kenzo, Ueno Takamasa
Joint Research Center for Human Retrovirus Infection, Kumamoto University, Kumamoto, Japan.
Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania.
Front Microbiol. 2021 Jul 7;12:703041. doi: 10.3389/fmicb.2021.703041. eCollection 2021.
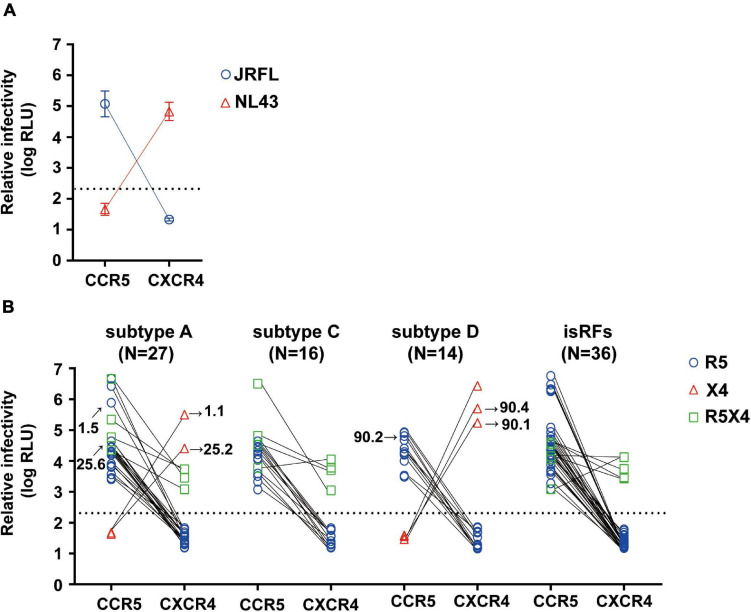
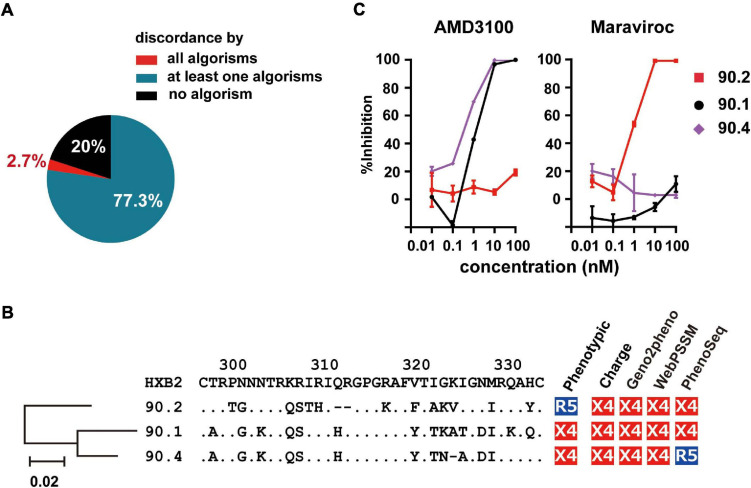
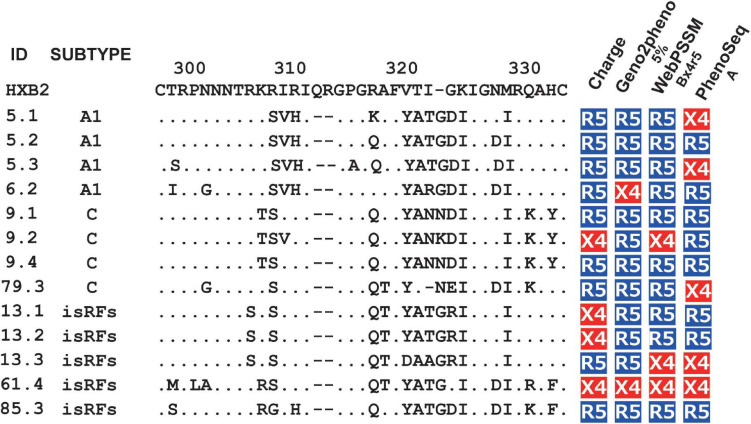
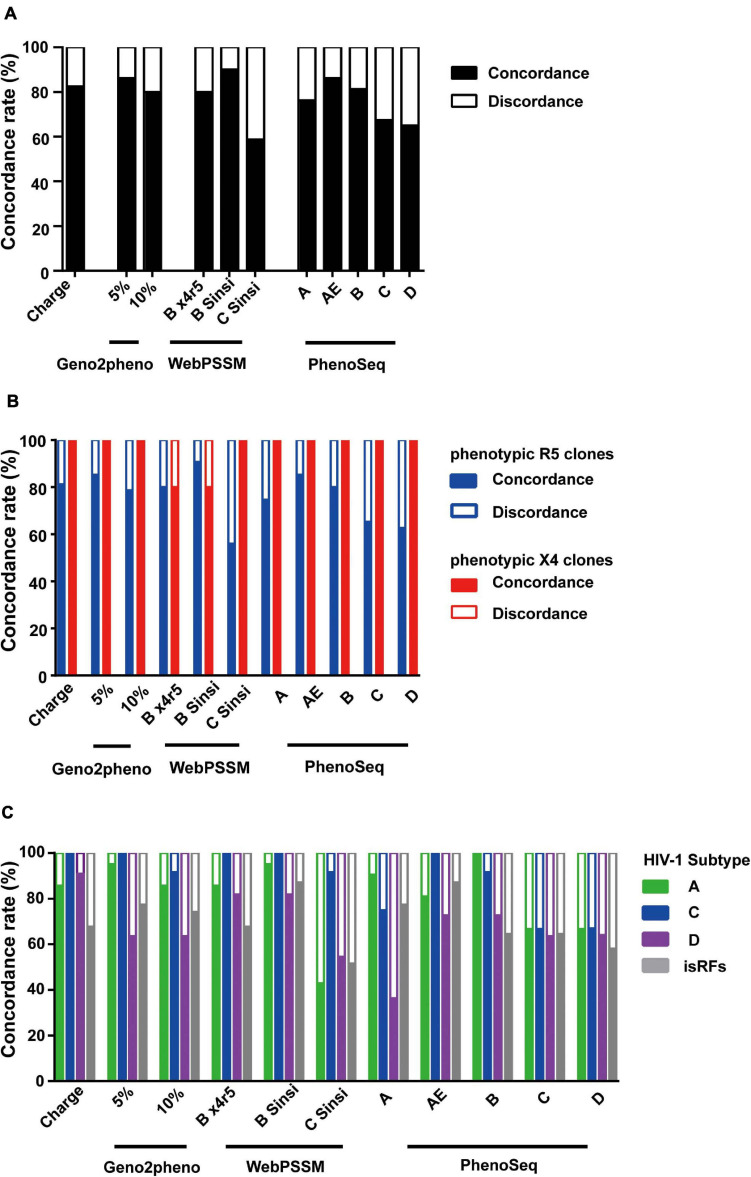
HIV human immunodeficiency virus type I (HIV-1) entry inhibitor potency is dependent on viral co-receptor tropisms and thereby tropism determination is clinically important. However, phenotypic tropisms of HIV-1 non-B subtypes have been poorly investigated and the genotypic prediction algorithms remain insufficiently validated. To clarify this issue, we recruited 52 treatment-naïve, HIV-1-infected patients in Tanzania, where multiple HIV-1 non-B subtypes co-circulate. Sequence analysis of 93 infectious envelope clones isolated from their plasma viral RNA revealed the co-circulation of subtypes A1, C, D, and inter-subtype recombinant forms (isRFs). Phenotypic tropism assays revealed that lentivirus reporters pseudotyped with 75 (80.6%) and 5 (5.4%) envelope clones could establish infection toward U87.CD4 cells expressing CCR5 (R5) and CXCR4 (X4), respectively; whereas the remaining 13 (14%) clones could infect both cells. Genotypic analyses by widely used algorithms including V3 net charge, Geno2pheno, WebPSSM, and PhenoSeq showed that almost all phenotypic X4-tropic clones and only 15 of 75 phenotypic R5-tropic clones were concordantly predicted. However, the remaining 60 phenotypic R5-tropic clones were discordantly predicted by at least one algorithm. In particular, 2 phenotypic R5-tropic clones were discordantly predicted by all algorithms tested. Taken together, the results demonstrate the limitation of currently available genotypic algorithms for predicting co-receptor inference among co-circulating multiple non-B subtypes and emerging isRFs. Also, the phenotypic tropism dataset presented here could be valuable for retraining of the widely used genotypic prediction algorithms to enhance their performance.
I型人类免疫缺陷病毒(HIV-1)进入抑制剂的效力取决于病毒共受体嗜性,因此嗜性测定在临床上具有重要意义。然而,对HIV-1非B亚型的表型嗜性研究较少,且基因型预测算法的验证仍不充分。为了阐明这一问题,我们在坦桑尼亚招募了52例未接受过治疗的HIV-1感染患者,该国多种HIV-1非B亚型共同流行。对从他们血浆病毒RNA中分离出的93个传染性包膜克隆进行序列分析,发现了A1、C、D亚型以及亚型间重组形式(isRFs)的共同流行。表型嗜性分析显示,用75个(80.6%)和5个(5.4%)包膜克隆假型化的慢病毒报告基因分别能够对表达CCR5(R5)和CXCR4(X4)的U87.CD4细胞建立感染;而其余13个(14%)克隆能够感染这两种细胞。通过包括V3净电荷、Geno2pheno、WebPSSM和PhenoSeq在内的广泛使用的算法进行基因型分析表明,几乎所有表型X4嗜性克隆以及75个表型R5嗜性克隆中只有15个被一致预测。然而,其余60个表型R5嗜性克隆至少被一种算法不一致地预测。特别是,2个表型R5嗜性克隆被所有测试算法不一致地预测。综上所述,结果表明目前可用的基因型算法在预测多种共同流行的非B亚型和新出现的isRFs之间的共受体推断方面存在局限性。此外,这里呈现的表型嗜性数据集对于重新训练广泛使用的基因型预测算法以提高其性能可能具有重要价值。