Janairo Gabriela Ilona B, Yu Derrick Ethelbhert C, Janairo Jose Isagani B
Chemistry Department, De La Salle University, 2401 Taft Avenue, 0922 Manila, Philippines.
Biology Department, De La Salle University, 2401 Taft Avenue, 0922 Manila, Philippines.
Netw Model Anal Health Inform Bioinform. 2021;10(1):51. doi: 10.1007/s13721-021-00326-2. Epub 2021 Jul 24.
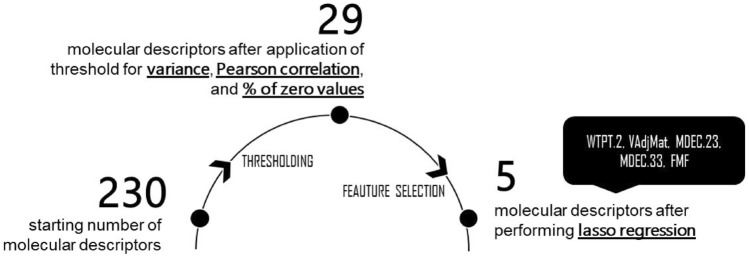
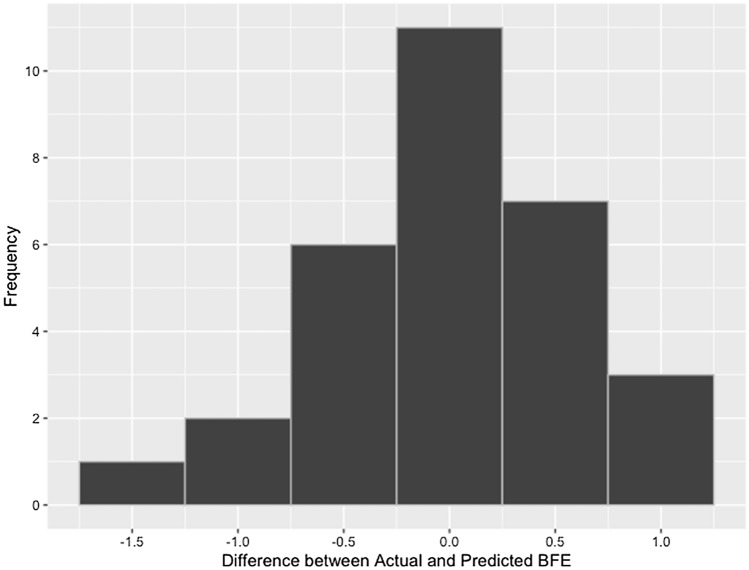
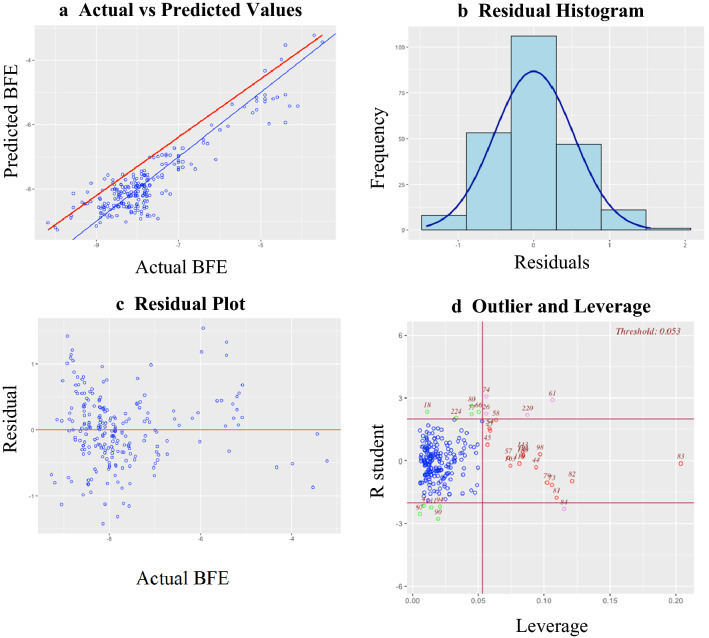
The widespread infection caused by the 2019 novel corona virus (SARS-CoV-2) has initiated global efforts to search for antiviral agents. Drug discovery is the first step in the development of commercially viable pharmaceutical products to deal with novel diseases. In an effort to accelerate the screening and drug discovery workflow for potential SARS-CoV-2 protease inhibitors, a machine learning model that can predict the binding free energies of compounds to the SARS-CoV-2 main protease is presented. The optimized multiple linear regression model, which was trained and tested on 226 natural compounds demonstrates reliable prediction performance ( test = 0.81, RMSE test = 0.43), while only requiring five topological descriptors. The externally validated model can help conserve and maximize available resources by limiting biological assays to compounds that yielded favorable outcomes from the model. The emergence of highly infectious diseases will always be a threat to human health and development, which is why the development of computational tools for rapid response is very important.
The online version contains supplementary material available at 10.1007/s13721-021-00326-2.
2019新型冠状病毒(SARS-CoV-2)引发的广泛感染促使全球展开对抗病毒药物的搜寻。药物研发是开发应对新型疾病的具有商业可行性的药品的第一步。为了加速潜在SARS-CoV-2蛋白酶抑制剂的筛选和药物研发流程,本文提出了一种能够预测化合物与SARS-CoV-2主要蛋白酶结合自由能的机器学习模型。该优化后的多元线性回归模型在226种天然化合物上进行了训练和测试,展现出可靠的预测性能(测试集R² = 0.81,测试集均方根误差RMSE = 0.43),且仅需五个拓扑描述符。经过外部验证的该模型能够通过将生物测定限制在模型预测结果良好的化合物上,有助于节省并最大化可用资源。高传染性疾病的出现始终是对人类健康和发展的威胁,这就是为何开发快速响应的计算工具非常重要的原因。
在线版本包含可在10.1007/s13721-021-00326-2获取的补充材料。