Department of Botany, Gauhati University, Guwahati, Assam, 781014, India.
Department of Molecular Biology and Biotechnology, Cotton University, Guwahati, Assam, 781001, India.
J Mol Model. 2021 Feb 28;27(3):97. doi: 10.1007/s00894-021-04703-6.
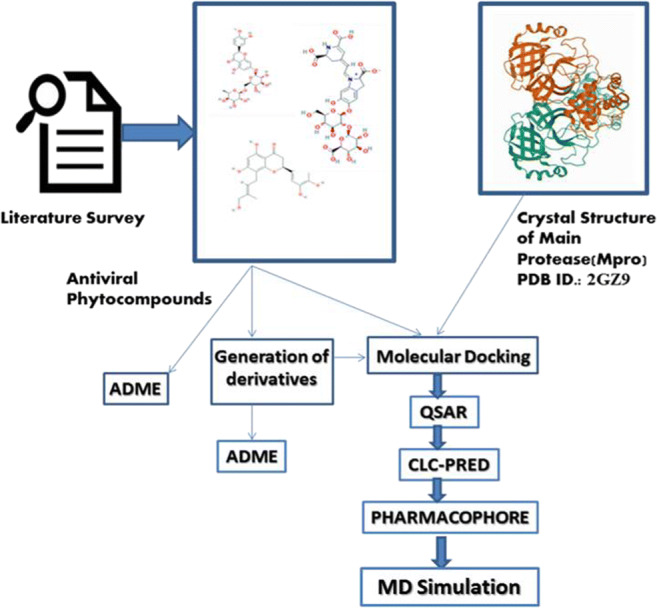
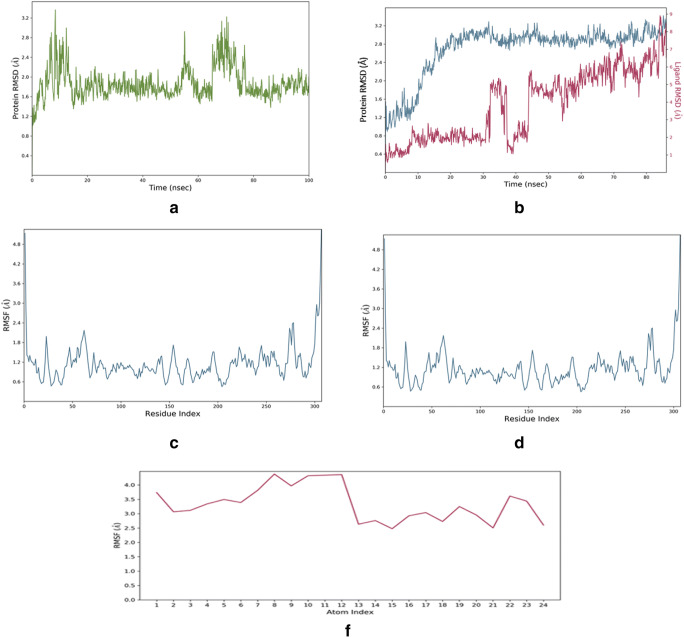
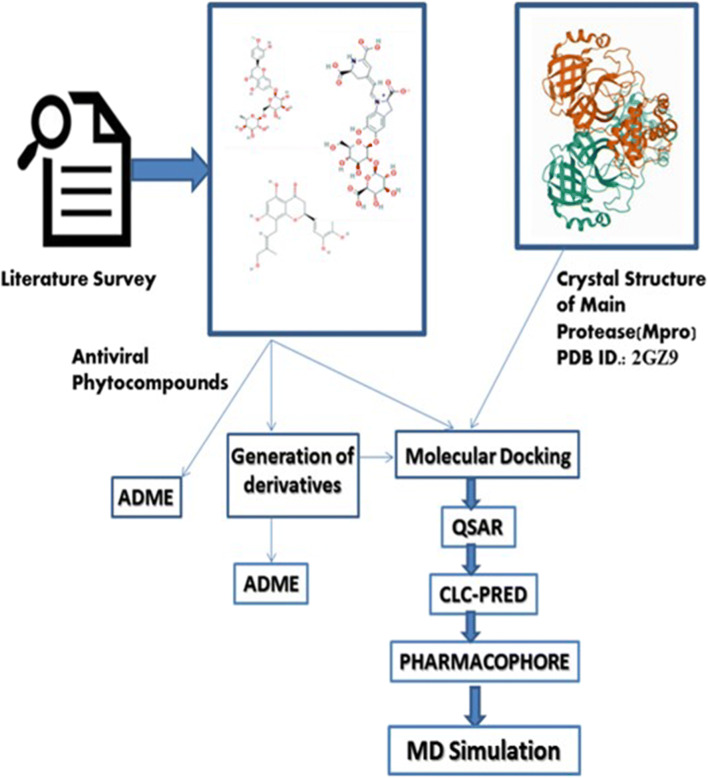
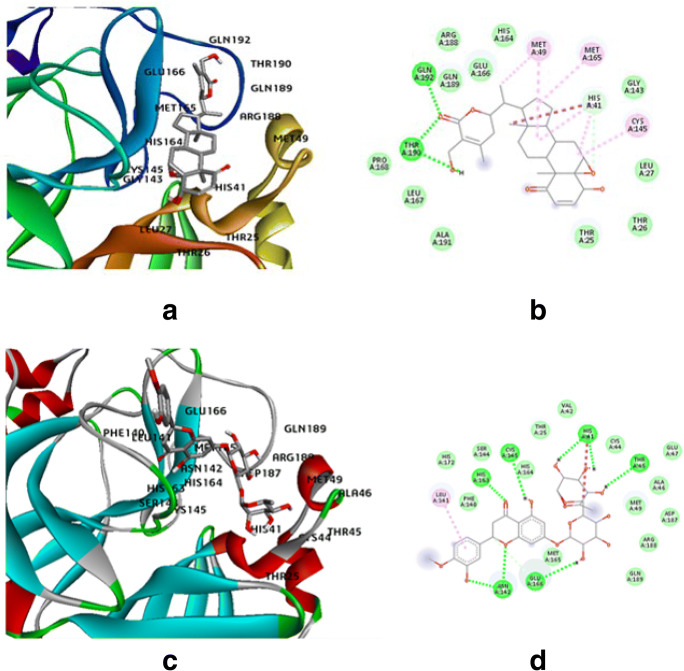
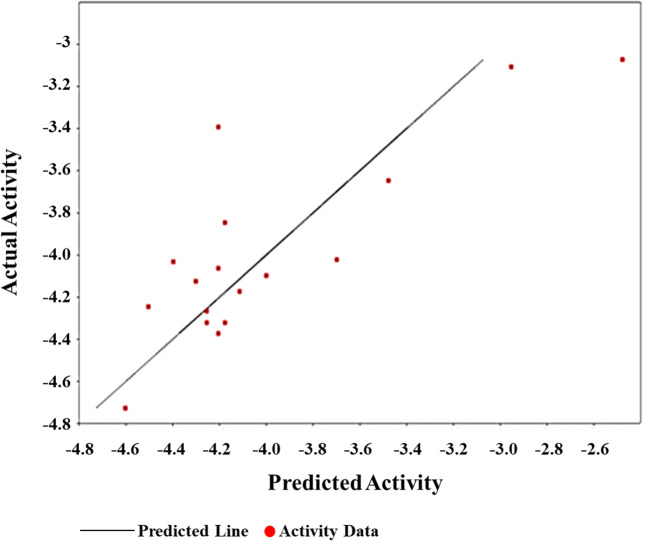
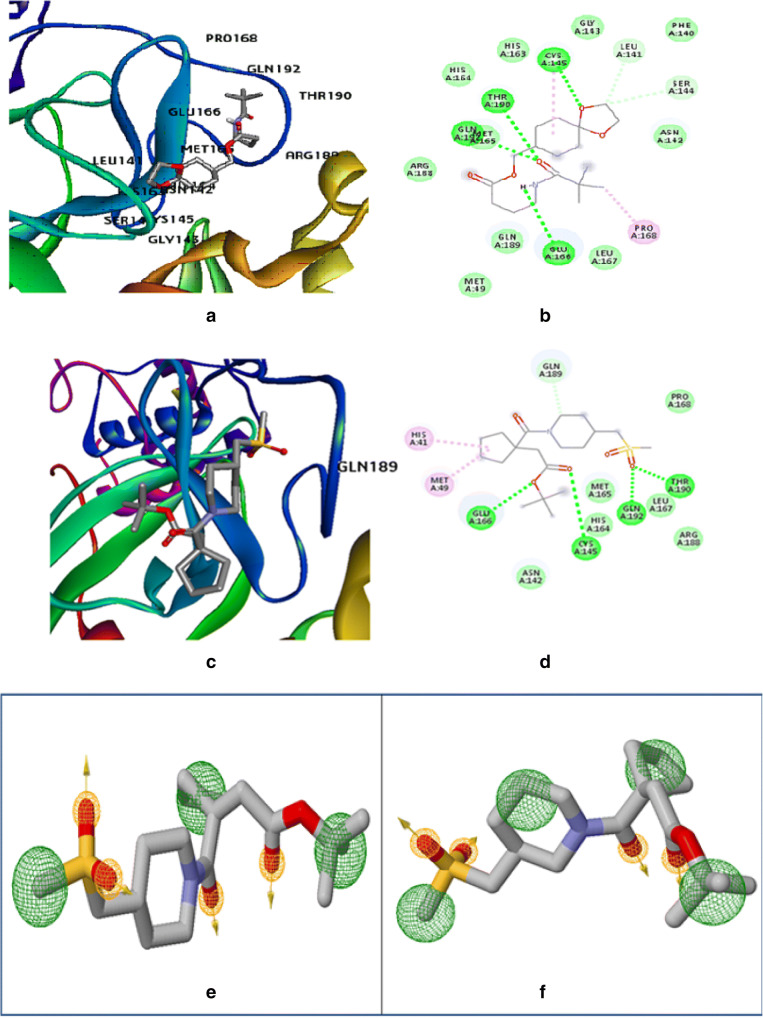
The spread of novel coronavirus SARS-CoV-2 has directed to a state of an unprecedented global pandemic. Many synthetic compounds and FDA-approved drugs have been significantly inhibitory against the virus, but no SARS-CoV-2 solution has been identified. However, small molecule fragment-based derivatives of potent phytocompounds may serve as promising inhibitors against SARS-CoV-2. In the pursuit of exploring novel SARS-CoV-2 inhibitors, we generated small molecule fragment derivatives from potent phytocompounds using neural networking and machine learning-based tools, which can cover unexplored regions of the chemical space that still retain lead-like properties. Out of 300 derivative molecules from withaferin-A, hesperidin, and baicalin, 30 were screened out with synthetic accessibility scores > 4 having the best ADME properties. The withaferin-A derivative molecules 61 and 64 exhibited a significant binding affinity of - 7.84 kcal/mol and - 7.94 kcal/mol. The docking study reveals that withaferin-A mol 61 forms 5 polar H-bonds with the M where amino acids involved are GLU166, THR190, CYS145, MET165, and GLN152 and upon QSAR analysis showed a minimal predicted IC50 value of 7762.47 nM. Furthermore, the in silico cytotoxicity predictions, pharmacophore modeling, and molecular dynamics simulation studies have resulted in predicting the highly potent small molecule derivative from withaferin-A (phytocompound from Withania somnifera) to be the potential inhibitor of SARS-CoV 2 protease (M) and a promising future lead candidate against COVID-19. The rationale of choosing withaferin-A from Withania somnifera (Ashwagandha) was propelled by the innumerous applications of Ashwagandha for the treatment of various antiviral diseases, common cold, and fever since time immemorial. Graphical abstract.
新型冠状病毒 SARS-CoV-2 的传播导致了前所未有的全球大流行。许多合成化合物和 FDA 批准的药物对该病毒具有显著的抑制作用,但尚未发现针对 SARS-CoV-2 的解决方案。然而,具有潜在药用价值的植物化合物的小分子片段衍生物可能成为有前途的 SARS-CoV-2 抑制剂。在探索新型 SARS-CoV-2 抑制剂的过程中,我们使用基于神经网络和机器学习的工具,从具有潜在药用价值的植物化合物中生成小分子片段衍生物,这些衍生物可以覆盖化学空间中尚未探索的区域,并且仍然具有类似先导的特性。在来自醉茄素-A、橙皮苷和黄芩苷的 300 个衍生分子中,有 30 个具有 >4 的合成可及性评分,具有最佳的 ADME 特性,被筛选出来。醉茄素-A 衍生物分子 61 和 64 表现出显著的结合亲和力,分别为 -7.84 kcal/mol 和 -7.94 kcal/mol。对接研究表明,醉茄素-A 分子 61 与 M 形成 5 个极性氢键,涉及的氨基酸为 GLU166、THR190、CYS145、MET165 和 GLN152,在 QSAR 分析中显示出最小预测 IC50 值为 7762.47 nM。此外,基于计算的细胞毒性预测、药效团建模和分子动力学模拟研究,预测出从醉茄素-A(来自印度人参的植物化合物)衍生而来的高活性小分子衍生物可能是 SARS-CoV 2 蛋白酶(M)的潜在抑制剂,也是针对 COVID-19 的有前途的未来先导候选药物。选择来自印度人参(印度人参)的醉茄素-A 的理由是,自古以来,印度人参就被广泛用于治疗各种抗病毒疾病、普通感冒和发烧。