Children's Hospital of Fudan University, Shanghai, China.
Department of Medicine, Austin Health, Epilepsy Research Centre, University of Melbourne, Heidelberg, Vic., Australia.
Epilepsia. 2021 Sep;62(9):2205-2217. doi: 10.1111/epi.17015. Epub 2021 Aug 2.
The classical description of Dravet syndrome, the prototypic developmental and epileptic encephalopathy, is of a normal 6-month-old infant presenting with a prolonged, febrile, hemiclonic seizure and showing developmental slowing after age 1 year. SCN1A pathogenic variants are found in >80% of patients. Many patients have atypical features resulting in diagnostic delay and inappropriate therapy. We aimed to provide an evidence-based definition of SCN1A-Dravet syndrome in readiness for precision medicine trials.
Epilepsy patients were recruited to the University of Melbourne Epilepsy Genetics Research Program between 1995 and 2020 by neurologists from around the world. Patients with SCN1A pathogenic variants were reviewed and only those with Dravet syndrome were included. Clinical data, including seizure and developmental course, were analyzed in all patients with SCN1A-Dravet syndrome.
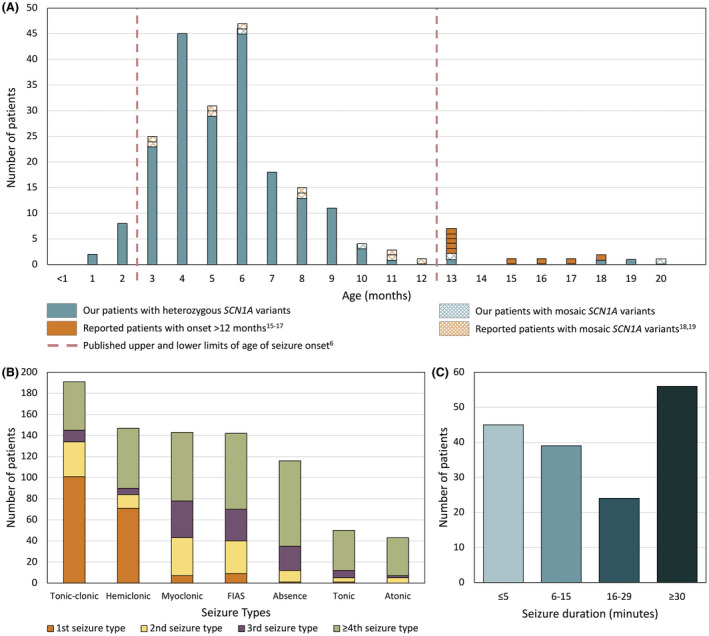
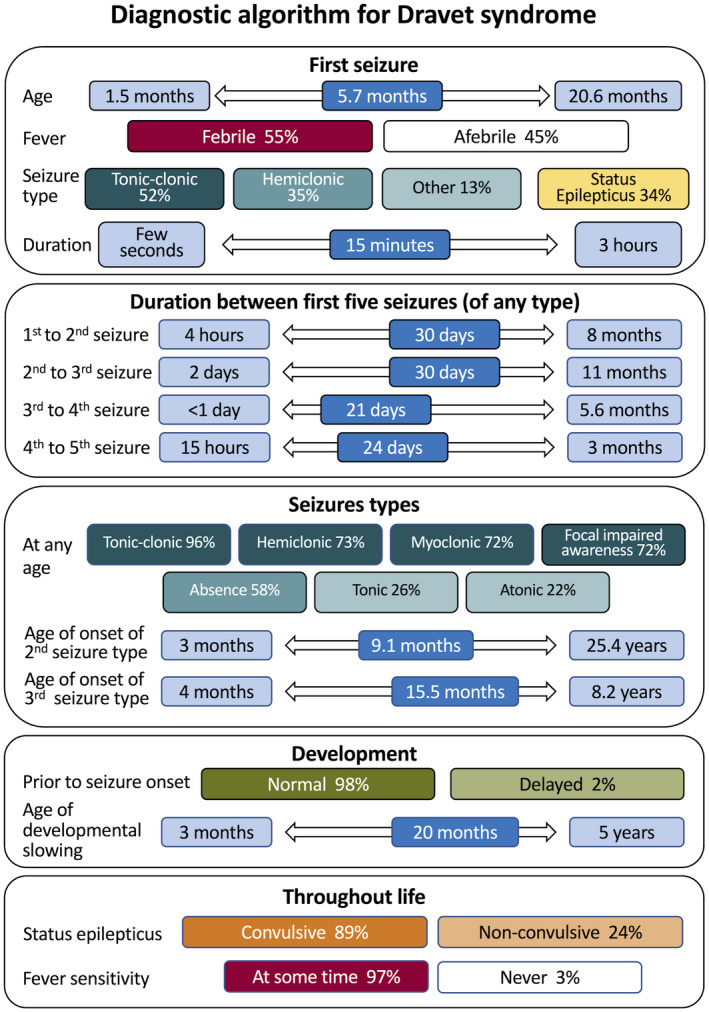
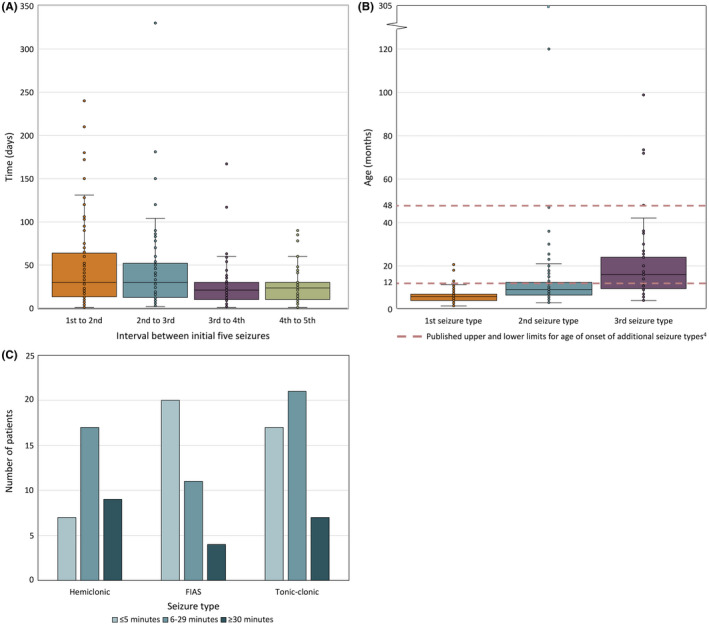
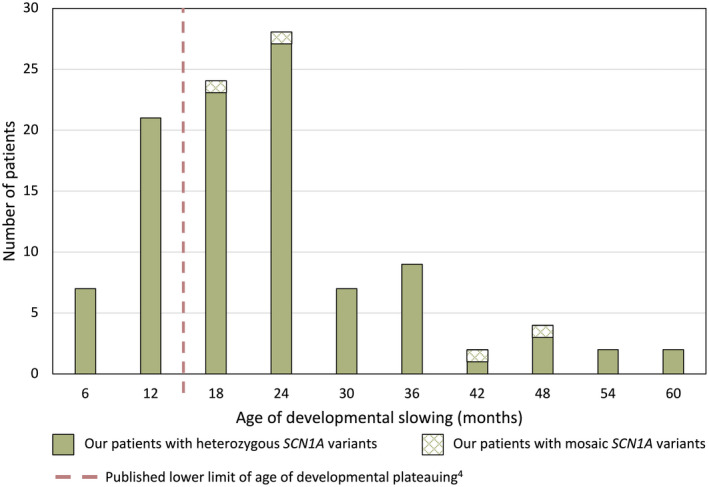
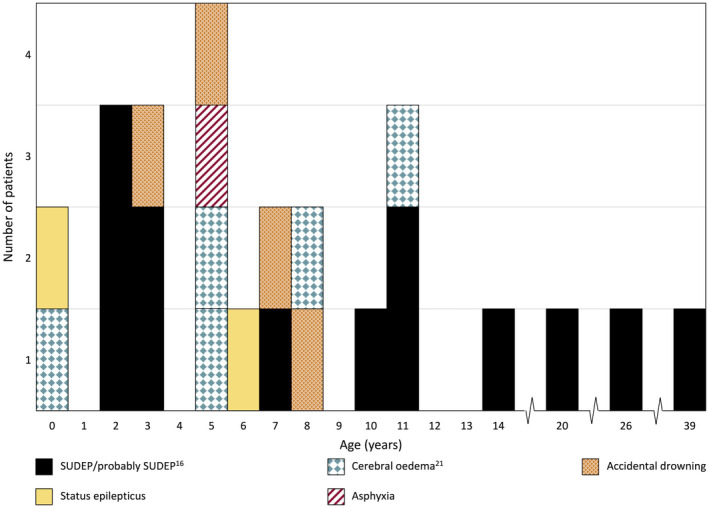
Two hundred and five patients were studied at a median age of 8.5 years (range 10 months to 60 years); 25 were deceased. The median seizure-onset age was 5.7 months (range 1.5-20.6 months). Initial seizures were tonic-clonic (52%) and hemiclonic (35%), with only 55% being associated with fever. Only 34% of patients presented with status epilepticus (seizure lasting ≥30 minutes). Median time between first and second seizure was 30 days (range 4 hours to 8 months), and seven patients (5%) had at least 6 months between initial seizures. Median ages at onset of second and third seizure types were 9.1 months (range 3 months-25.4 years) and 15.5 months (range 4 months-8.2 years), respectively. Developmental slowing occurred prior to 12 months in 27%.
An evidence-based definition of SCN1A-Dravet syndrome is essential for early diagnosis. We refine the spectrum of Dravet syndrome, based on patterns of seizure onset, type, and progression. Understanding of the full spectrum of SCN1A-Dravet syndrome presentation is essential for early diagnosis and optimization of treatment, especially as precision medicine trials become available.
德拉维特综合征(Dravet syndrome)是一种典型的发育性癫痫性脑病,其经典描述为 6 月龄正常婴儿出现长时间发热性、半侧阵挛性癫痫发作,且 1 岁后出现发育迟缓。80%以上的患者存在 SCN1A 致病性变异。许多患者存在非典型特征,导致诊断延迟和治疗不当。我们旨在提供 SCN1A 德拉维特综合征的循证定义,为精准医学试验做好准备。
1995 年至 2020 年间,来自世界各地的神经科医生通过全球癫痫遗传学研究计划招募了癫痫患者。对 SCN1A 致病性变异患者进行了回顾性研究,仅纳入具有德拉维特综合征的患者。对所有 SCN1A 德拉维特综合征患者进行临床数据(包括发作和发育过程)分析。
研究共纳入 205 例患者,中位年龄 8.5 岁(范围 10 个月至 60 岁);25 例患者死亡。中位发病年龄为 5.7 个月(范围 1.5-20.6 个月)。首发癫痫发作形式为强直-阵挛性(52%)和半侧阵挛性(35%),仅 55%与发热相关。仅有 34%的患者出现癫痫持续状态(发作持续≥30 分钟)。首次发作和第二次发作之间的中位时间为 30 天(范围 4 小时至 8 个月),7 例患者(5%)在首次发作后至少 6 个月出现第二次发作。第二次和第三次发作类型的中位发病年龄分别为 9.1 个月(范围 3 个月至 25.4 岁)和 15.5 个月(范围 4 个月至 8.2 岁)。27%的患者在 12 个月前出现发育迟缓。
SCN1A 德拉维特综合征的循证定义对于早期诊断至关重要。我们根据癫痫发作起始、类型和进展模式,对德拉维特综合征的谱进行了细化。全面了解 SCN1A 德拉维特综合征的表现对于早期诊断和优化治疗至关重要,尤其是随着精准医学试验的开展。