Chen Hongjie, Majumdar Arunabha, Wang Lu, Kar Siddhartha, Brown Kevin M, Feng Helian, Turman Constance, Dennis Joe, Easton Douglas, Michailidou Kyriaki, Simard Jacques, Bishop Timothy, Cheng Iona C, Huyghe Jeroen R, Schmit Stephanie L, O'Mara Tracy A, Spurdle Amanda B, Gharahkhani Puya, Schumacher Johannes, Jankowski Janusz, Gockel Ines, Bondy Melissa L, Houlston Richard S, Jenkins Robert B, Melin Beatrice, Lesseur Corina, Ness Andy R, Diergaarde Brenda, Olshan Andrew F, Amos Christopher I, Christiani David C, Landi Maria T, McKay James D, Brossard Myriam, Iles Mark M, Law Matthew H, MacGregor Stuart, Beesley Jonathan, Jones Michelle R, Tyrer Jonathan, Winham Stacey J, Klein Alison P, Petersen Gloria, Li Donghui, Wolpin Brian M, Eeles Rosalind A, Haiman Christopher A, Kote-Jarai Zsofia, Schumacher Fredrick R, Brennan Paul, Chanock Stephen J, Gaborieau Valerie, Purdue Mark P, Pharoah Paul, Hung Rayjean J, Amundadottir Laufey T, Kraft Peter, Pasaniuc Bogdan, Lindström Sara
Department of Epidemiology, University of Washington, Seattle, WA, USA.
Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA.
HGG Adv. 2021 Jul 8;2(3):100041. doi: 10.1016/j.xhgg.2021.100041. Epub 2021 Jun 12.
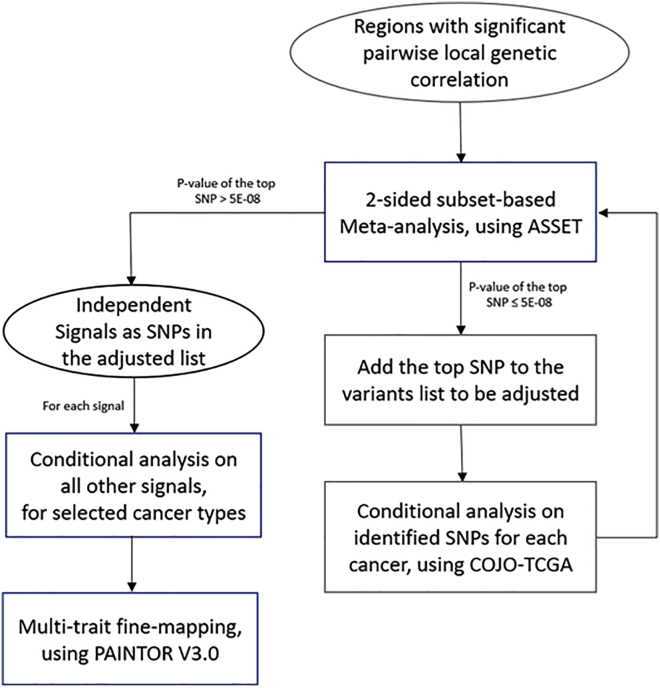
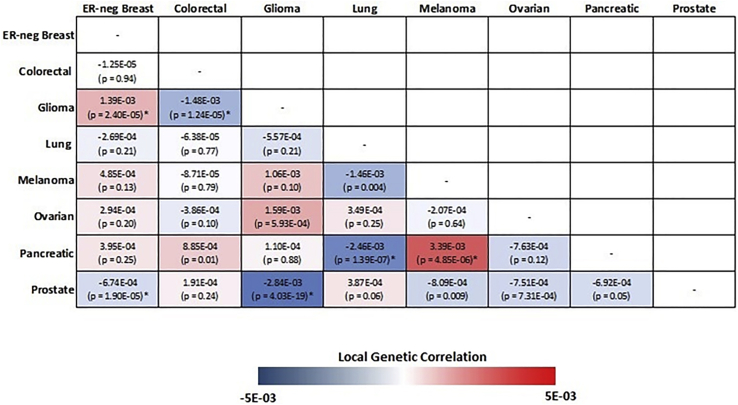
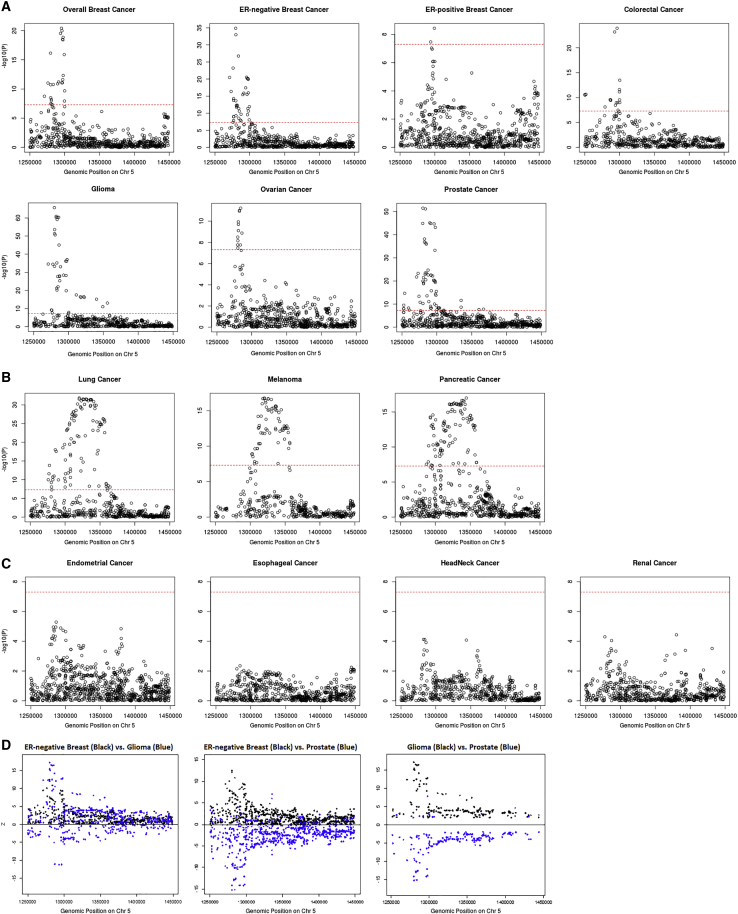
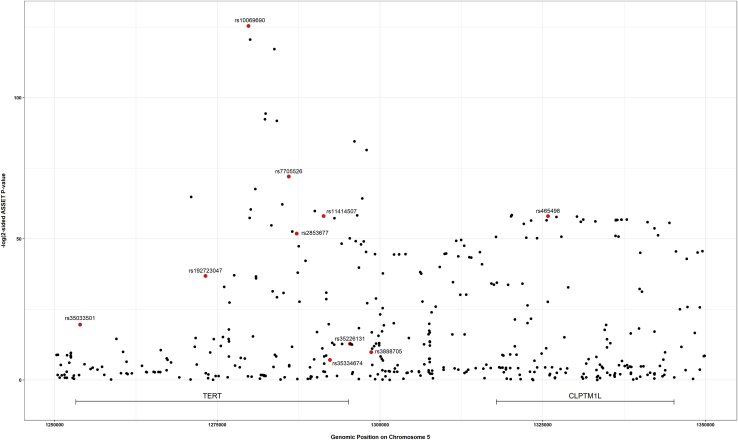
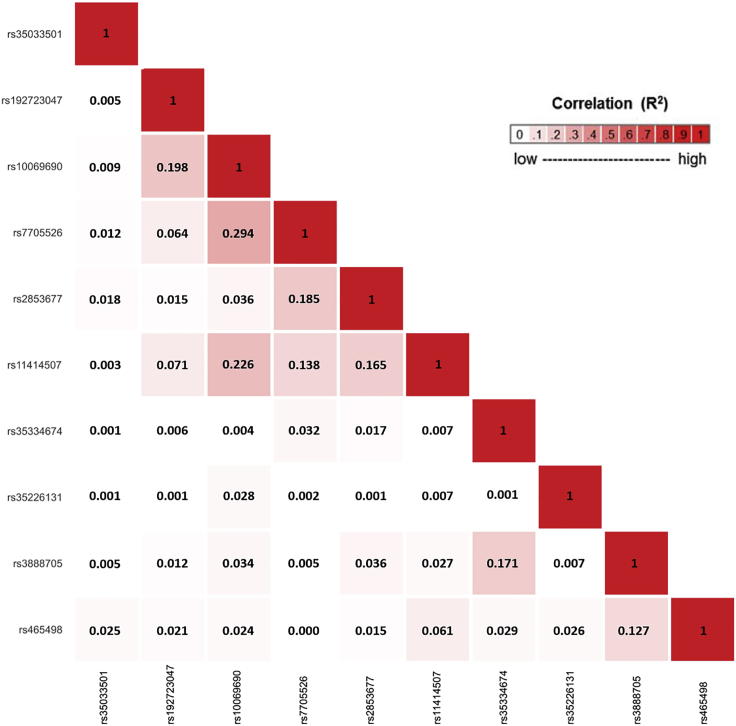
Genome-wide association studies (GWASs) have identified thousands of cancer risk loci revealing many risk regions shared across multiple cancers. Characterizing the cross-cancer shared genetic basis can increase our understanding of global mechanisms of cancer development. In this study, we collected GWAS summary statistics based on up to 375,468 cancer cases and 530,521 controls for fourteen types of cancer, including breast (overall, estrogen receptor [ER]-positive, and ER-negative), colorectal, endometrial, esophageal, glioma, head/neck, lung, melanoma, ovarian, pancreatic, prostate, and renal cancer, to characterize the shared genetic basis of cancer risk. We identified thirteen pairs of cancers with statistically significant local genetic correlations across eight distinct genomic regions. Specifically, the 5p15.33 region, harboring the and genes, showed statistically significant local genetic correlations for multiple cancer pairs. We conducted a cross-cancer fine-mapping of the 5p15.33 region based on eight cancers that showed genome-wide significant associations in this region (ER-negative breast, colorectal, glioma, lung, melanoma, ovarian, pancreatic, and prostate cancer). We used an iterative analysis pipeline implementing a subset-based meta-analysis approach based on cancer-specific conditional analyses and identified ten independent cross-cancer associations within this region. For each signal, we conducted cross-cancer fine-mapping to prioritize the most plausible causal variants. Our findings provide a more in-depth understanding of the shared inherited basis across human cancers and expand our knowledge of the 5p15.33 region in carcinogenesis.
全基因组关联研究(GWAS)已鉴定出数千个癌症风险位点,揭示了多种癌症共有的许多风险区域。表征跨癌症共享的遗传基础可以增进我们对癌症发展全球机制的理解。在本研究中,我们收集了基于多达375468例癌症病例和530521例对照的GWAS汇总统计数据,涉及十四种癌症,包括乳腺癌(总体、雌激素受体[ER]阳性和ER阴性)、结直肠癌、子宫内膜癌、食管癌、神经胶质瘤、头颈癌、肺癌、黑色素瘤、卵巢癌、胰腺癌、前列腺癌和肾癌,以表征癌症风险的共享遗传基础。我们在八个不同的基因组区域中鉴定出十三对具有统计学显著局部遗传相关性的癌症。具体而言,包含 和 基因的5p15.33区域在多对癌症中显示出统计学显著的局部遗传相关性。我们基于在该区域显示全基因组显著关联的八种癌症(ER阴性乳腺癌、结直肠癌、神经胶质瘤、肺癌、黑色素瘤、卵巢癌、胰腺癌和前列腺癌)对5p15.33区域进行了跨癌症精细定位。我们使用了一种迭代分析流程,该流程基于癌症特异性条件分析实施基于子集的荟萃分析方法,并在该区域内鉴定出十个独立的跨癌症关联。对于每个信号,我们进行了跨癌症精细定位,以对最可能的因果变异进行优先级排序。我们的发现为人类癌症共享的遗传基础提供了更深入的理解,并扩展了我们对5p15.33区域在致癌作用中的认识。