Martinez-Peinado Nieves, Lorente-Macías Álvaro, García-Salguero Alejandro, Cortes-Serra Nuria, Fenollar-Collado Ángel, Ros-Lucas Albert, Gascon Joaquim, Pinazo Maria-Jesus, Molina Ignacio J, Unciti-Broceta Asier, Díaz-Mochón Juan J, Pineda de Las Infantas Y Villatoro María J, Izquierdo Luis, Alonso-Padilla Julio
Barcelona Institute for Global Health (ISGlobal), Hospital Clínic-University of Barcelona, 08036 Barcelona, Spain.
Department of Medicinal & Organic Chemistry and Excellence Research Unit of "Chemistry Applied to Biomedicine and the Environment", Faculty of Pharmacy, University of Granada, Campus de Cartuja s/n, 18071 Granada, Spain.
Pharmaceuticals (Basel). 2021 Jul 1;14(7):638. doi: 10.3390/ph14070638.
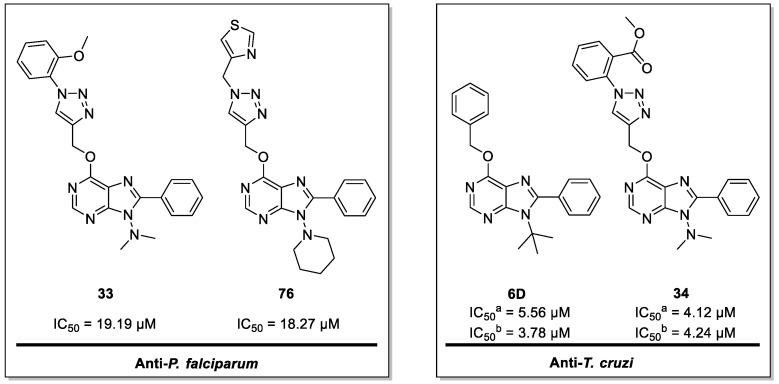
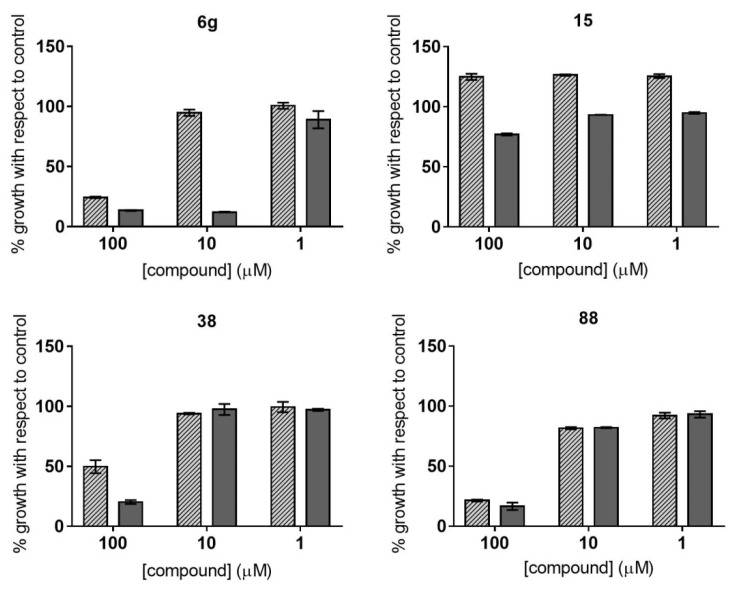
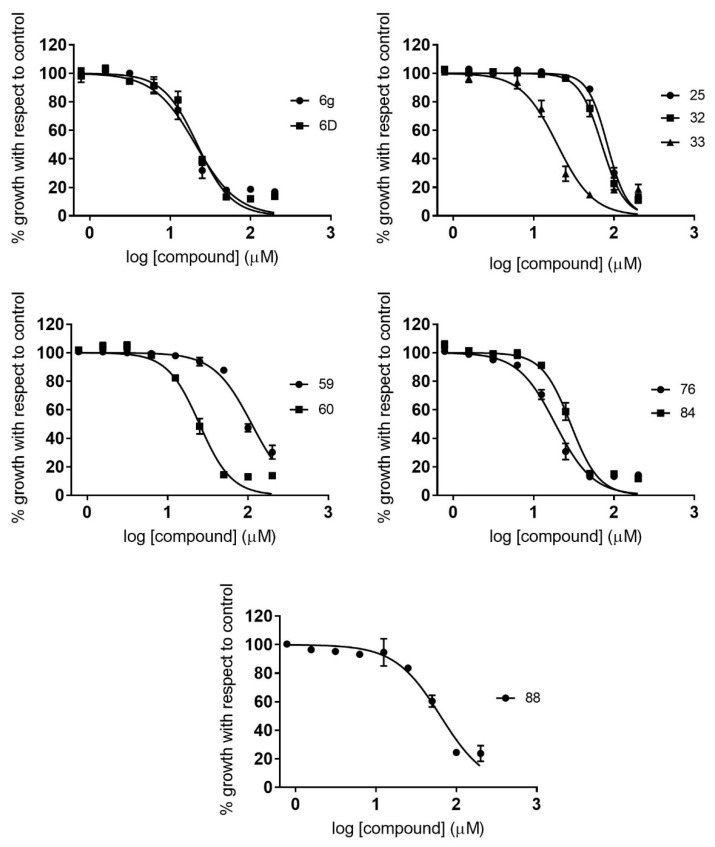
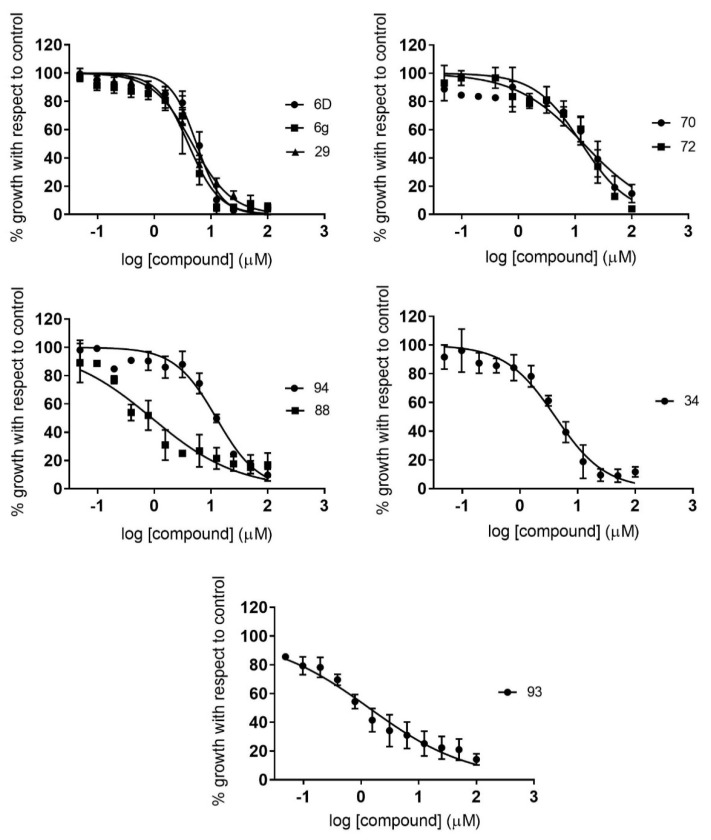
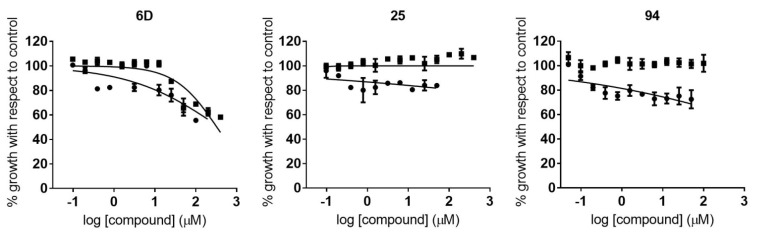
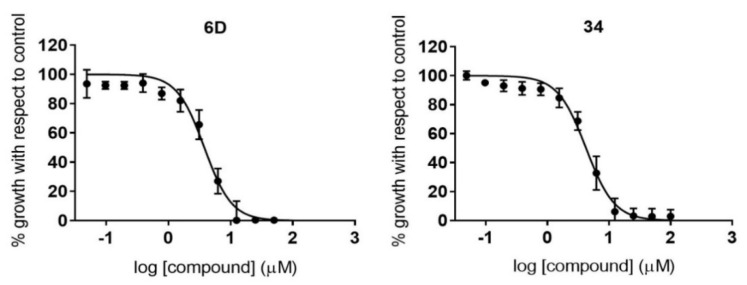
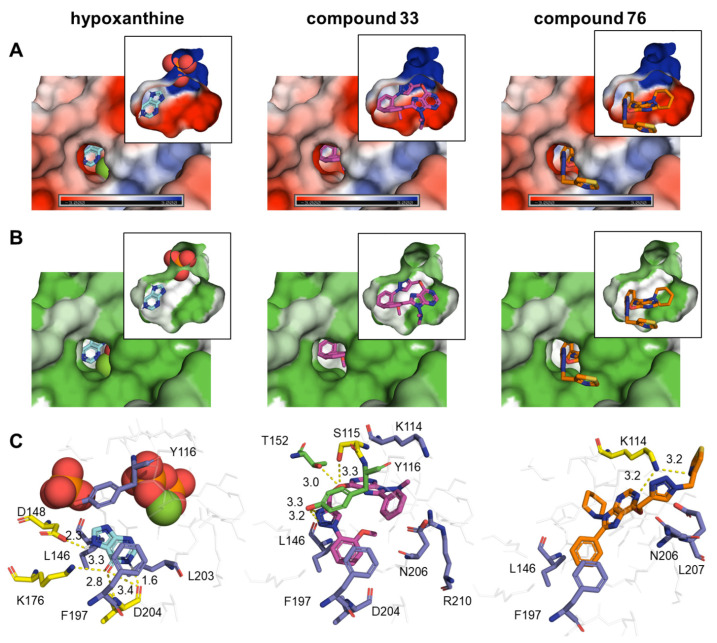
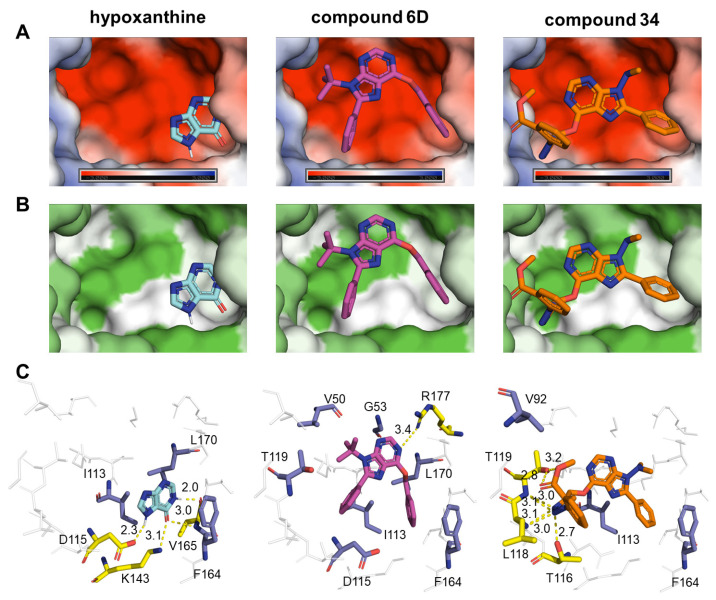
Malaria and Chagas disease, caused by spp. and parasites, remain important global health problems. Available treatments for those diseases present several limitations, such as lack of efficacy, toxic side effects, and drug resistance. Thus, new drugs are urgently needed. The discovery of new drugs may be benefited by considering the significant biological differences between hosts and parasites. One of the most striking differences is found in the purine metabolism, because most of the parasites are incapable of de novo purine biosynthesis. Herein, we have analyzed the in vitro anti- and anti- activity of a collection of 81 purine derivatives and pyrimidine analogs. We firstly used a primary screening at three fixed concentrations (100, 10, and 1 µM) and progressed those compounds that kept the growth of the parasites < 30% at 100 µM to dose-response assays. Then, we performed two different cytotoxicity assays on Vero cells and human HepG2 cells. Finally, compounds specifically active against were tested against intracellular amastigote forms. Purines (IC = 19.19 µM) and (IC = 18.27 µM) were the most potent against . On the other hand, (IC = 3.78 µM) and (IC = 4.24 µM) were identified as hit purines against amastigotes. Moreover, an in silico docking study revealed that and hypoxanthine guanine phosphoribosyltransferase enzymes could be the potential targets of those compounds. Our study identified two novel, purine-based chemotypes that could be further optimized to generate potent and diversified anti-parasitic drugs against both parasites.
由疟原虫属物种和克氏锥虫寄生虫引起的疟疾和恰加斯病仍然是重要的全球健康问题。针对这些疾病的现有治疗方法存在若干局限性,如疗效不佳、毒副作用和耐药性。因此,迫切需要新药。考虑到宿主和寄生虫之间显著的生物学差异,可能有助于发现新药。最显著的差异之一存在于嘌呤代谢中,因为大多数寄生虫无法进行嘌呤的从头生物合成。在此,我们分析了81种嘌呤衍生物和嘧啶类似物的体外抗疟原虫和抗克氏锥虫活性。我们首先在三个固定浓度(100、10和1 μM)下进行初步筛选,并将在100 μM时使寄生虫生长保持在< 30%的那些化合物推进到剂量反应测定中。然后,我们对非洲绿猴肾细胞和人肝癌细胞系HepG2细胞进行了两种不同的细胞毒性测定。最后,针对对克氏锥虫具有特异性活性的化合物对细胞内无鞭毛体形式进行了测试。嘌呤化合物P1(IC50 = 19.19 μM)和P2(IC50 = 18.27 μM)对疟原虫最有效。另一方面,P3(IC50 = 3.78 μM)和P4(IC50 = 4.24 μM)被确定为针对克氏锥虫无鞭毛体的有效嘌呤化合物。此外,一项计算机对接研究表明,P1和P2与次黄嘌呤鸟嘌呤磷酸核糖转移酶可能是这些化合物的潜在靶点。我们的研究确定了两种新型的基于嘌呤的化学类型,可进一步优化以产生针对这两种寄生虫的强效且多样化的抗寄生虫药物。