Institut Cochin, Inserm U1016, CNRS UMR 8104, Paris Descartes University UMR-S1016, 75014, Paris, France.
INSERM UMR1170, Equipe Labellisée Ligue Nationale Contre Le Cancer, Gustave Roussy, Paris-Saclay University, 94800, Villejuif, France.
BMC Bioinformatics. 2021 Aug 17;22(1):407. doi: 10.1186/s12859-021-04320-3.
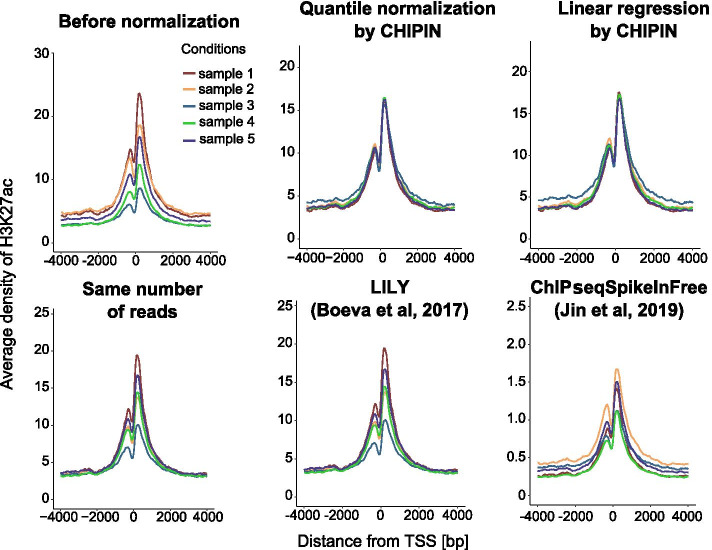
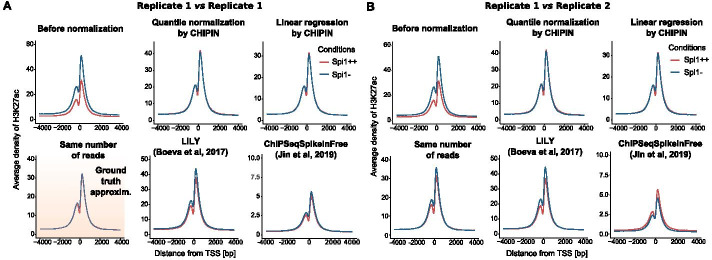
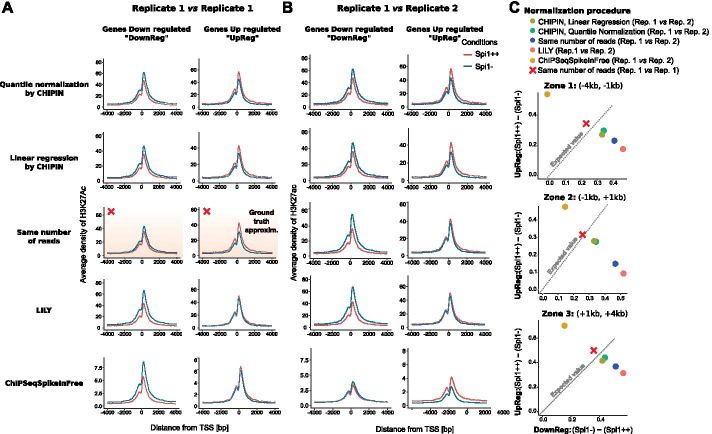
Multiple studies rely on ChIP-seq experiments to assess the effect of gene modulation and drug treatments on protein binding and chromatin structure. However, most methods commonly used for the normalization of ChIP-seq binding intensity signals across conditions, e.g., the normalization to the same number of reads, either assume a constant signal-to-noise ratio across conditions or base the estimates of correction factors on genomic regions with intrinsically different signals between conditions. Inaccurate normalization of ChIP-seq signal may, in turn, lead to erroneous biological conclusions.
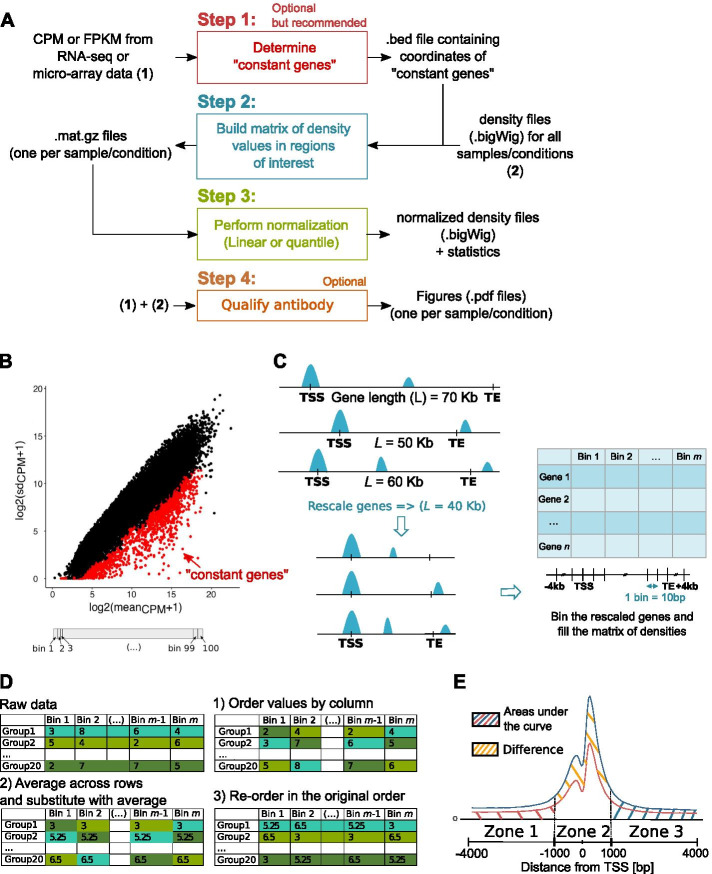
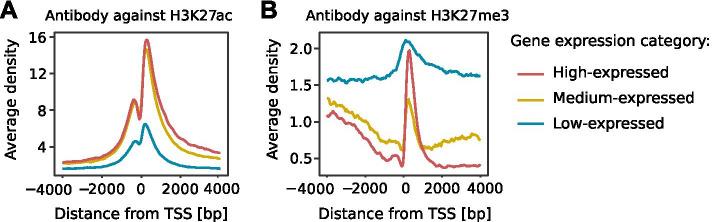
We developed a new R package, CHIPIN, that allows normalizing ChIP-seq signals across different conditions/samples when spike-in information is not available, but gene expression data are at hand. Our normalization technique is based on the assumption that, on average, no differences in ChIP-seq signals should be observed in the regulatory regions of genes whose expression levels are constant across samples/conditions. In addition to normalizing ChIP-seq signals, CHIPIN provides as output a number of graphs and calculates statistics allowing the user to assess the efficiency of the normalization and qualify the specificity of the antibody used. In addition to ChIP-seq, CHIPIN can be used without restriction on open chromatin ATAC-seq or DNase hypersensitivity data. We validated the CHIPIN method on several ChIP-seq data sets and documented its superior performance in comparison to several commonly used normalization techniques.
The CHIPIN method provides a new way for ChIP-seq signal normalization across conditions when spike-in experiments are not available. The method is implemented in a user-friendly R package available on GitHub: https://github.com/BoevaLab/CHIPIN.
多项研究依赖于 ChIP-seq 实验来评估基因调控和药物处理对蛋白质结合和染色质结构的影响。然而,大多数用于跨条件归一化 ChIP-seq 结合强度信号的常用方法,例如,归一化为相同数量的读数,要么假设条件之间的信号噪声比恒定,要么基于条件之间内在信号不同的基因组区域来估计校正因子。ChIP-seq 信号的归一化不准确反过来可能导致错误的生物学结论。
我们开发了一个新的 R 包 CHIPIN,当没有 Spike-in 信息可用但手头有基因表达数据时,它允许在不同条件/样本之间归一化 ChIP-seq 信号。我们的归一化技术基于这样的假设,即在平均情况下,在基因的调控区域中不应该观察到 ChIP-seq 信号的差异,这些基因的表达水平在样本/条件之间是恒定的。除了归一化 ChIP-seq 信号外,CHIPIN 还提供了许多图形和计算统计信息,允许用户评估归一化的效率并确定所用抗体的特异性。除了 ChIP-seq 之外,CHIPIN 还可以不受限制地用于开放染色质 ATAC-seq 或 DNase 超敏数据。我们在几个 ChIP-seq 数据集上验证了 CHIPIN 方法,并记录了与几种常用归一化技术相比,该方法的优越性能。
当没有 Spike-in 实验时,CHIPIN 方法为跨条件的 ChIP-seq 信号归一化提供了一种新方法。该方法已在 GitHub 上的用户友好的 R 包中实现:https://github.com/BoevaLab/CHIPIN。