National Engineering Laboratory for Druggable Gene and Protein Screening, Northeast Normal University, Changchun, China.
Center of Genome Analysis, ABLife BioBigData Institute, Wuhan, China.
Front Immunol. 2021 Aug 20;12:644350. doi: 10.3389/fimmu.2021.644350. eCollection 2021.
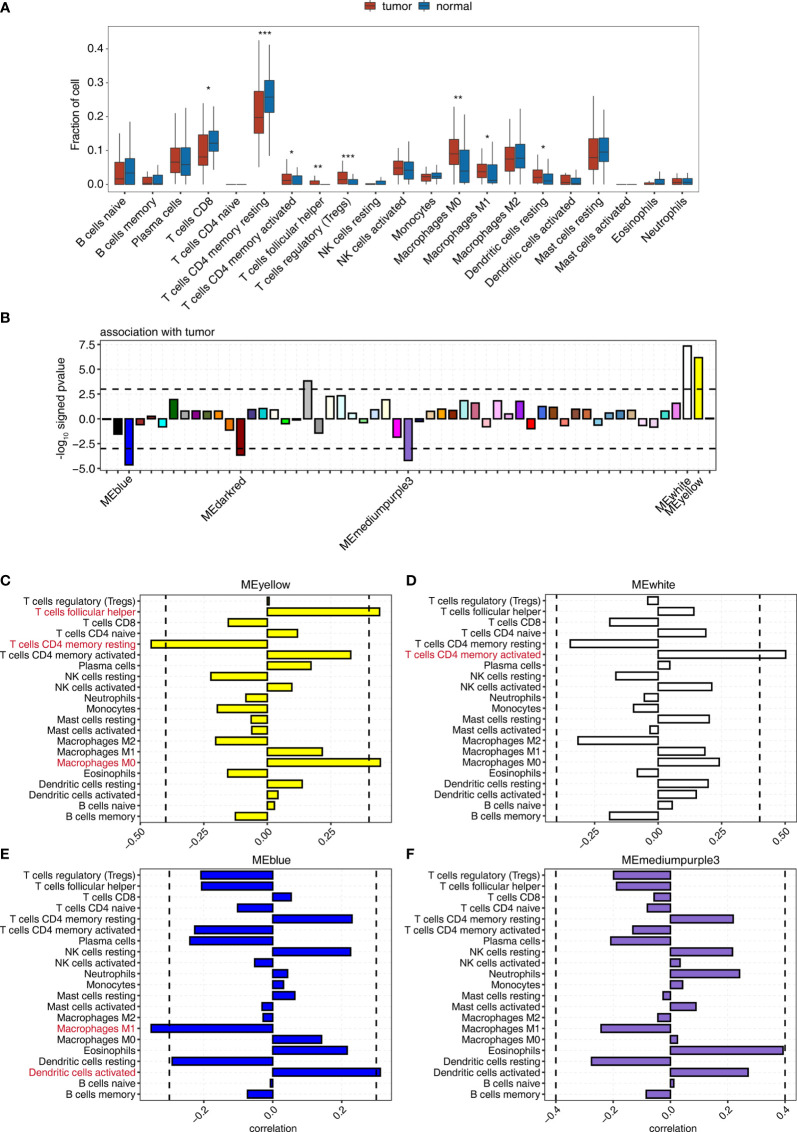
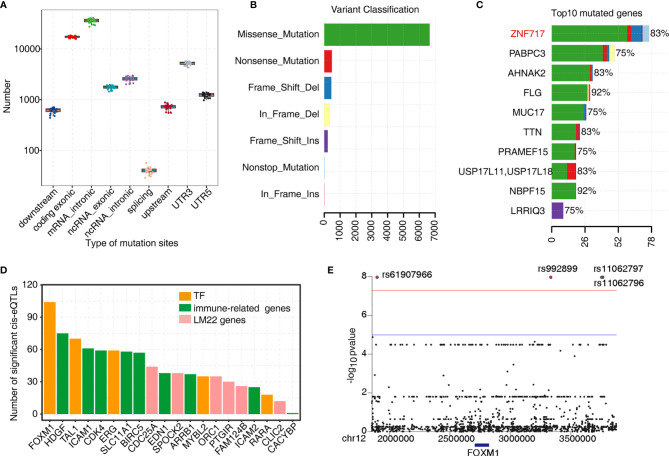
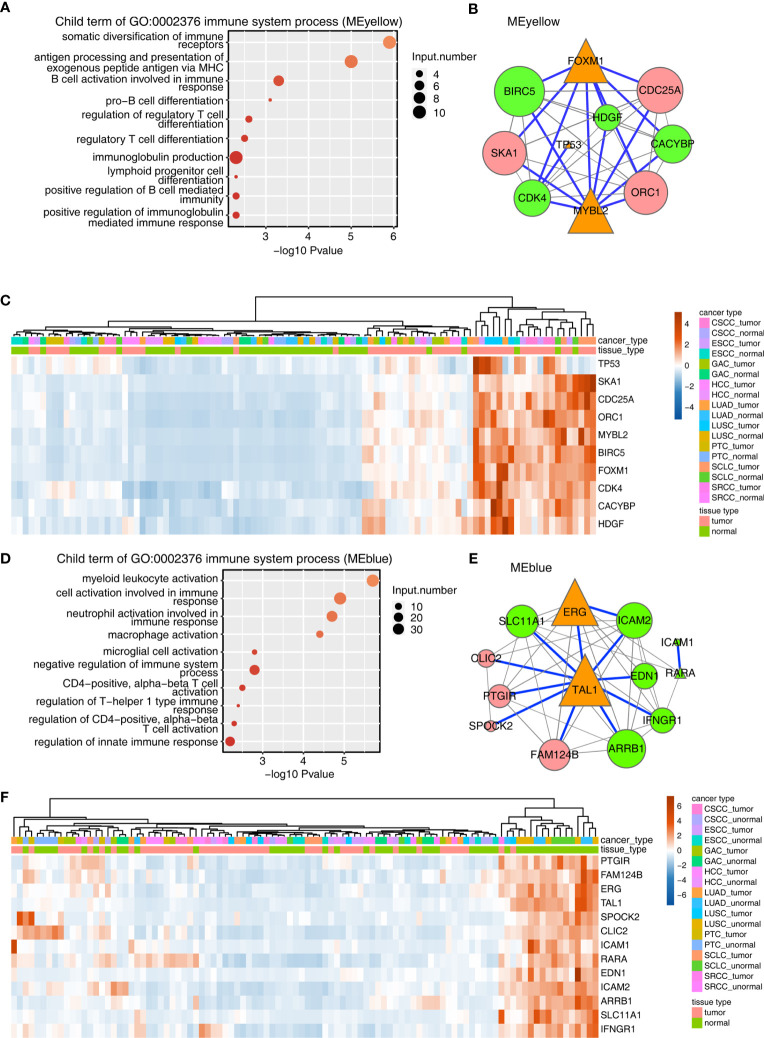
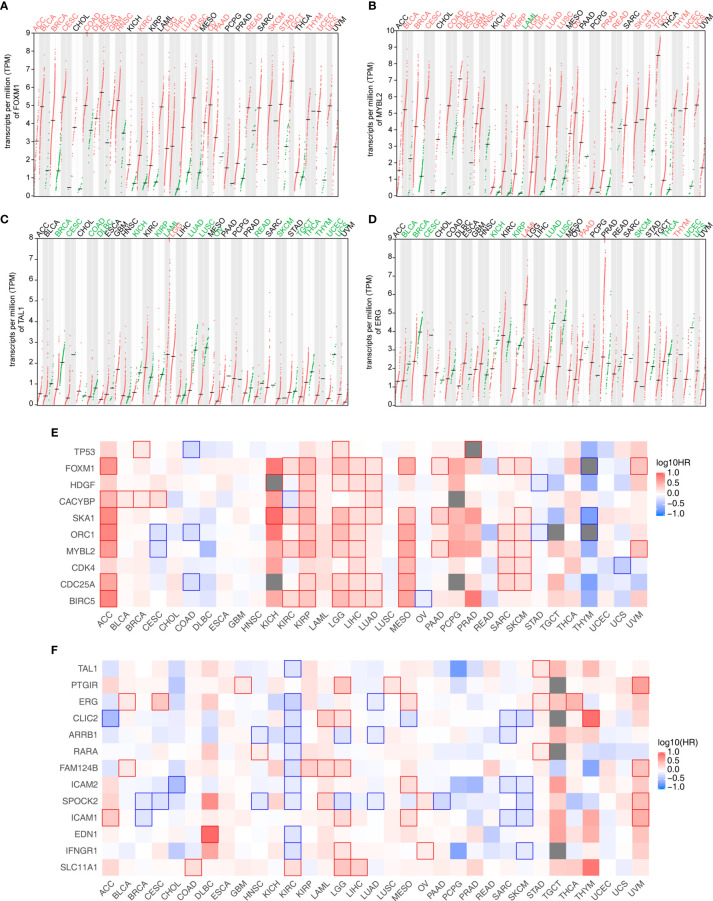
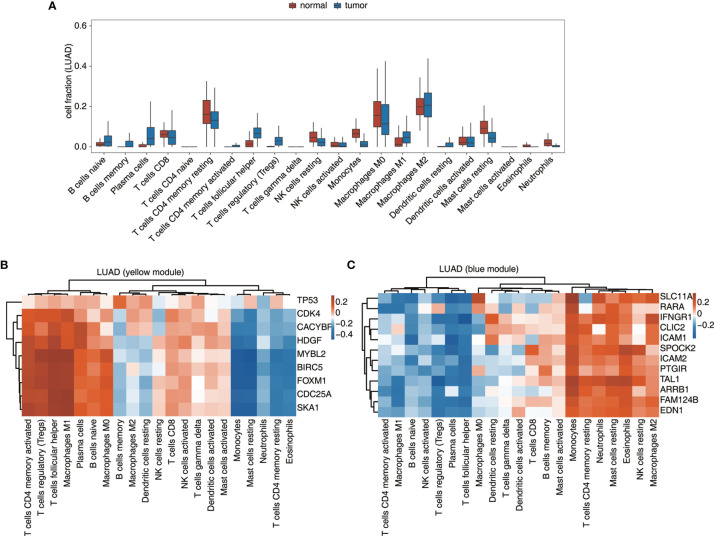
Tumor-infiltrating immune cells shape the tumor microenvironment and are closely related to clinical outcomes. Several transcription factors (TFs) have also been reported to regulate the antitumor activity and immune cell infiltration. This study aimed to quantify the populations of different immune cells infiltrated in tumor samples based on the bulk RNA sequencing data obtained from 50 cancer patients using the CIBERSORT and the EPIC algorithm. Weighted gene coexpression network analysis (WGCNA) identified eigengene modules strongly associated with tumorigenesis and the activation of CD4+ memory T cells, dendritic cells, and macrophages. TF genes , , , and are central in the subnetworks of the eigengene modules associated with immune-related genes. The analysis of The Cancer Genome Atlas (TCGA) cancer data confirmed these findings and further showed that the expression of these potential TF genes regulating immune infiltration, and the immune-related genes that they regulated, was associated with the survival of patients within multiple cancers. Exome-seq was performed on 24 paired samples that also had RNA-seq data. The expression quantitative trait loci (eQTL) analysis showed that mutations were significantly more frequent in the regions flanking the TF genes compared with those of non-TF genes, suggesting a driver role of these TF genes regulating immune infiltration. Taken together, this study presented a practical method for identifying genes that regulate immune infiltration. These genes could be potential biomarkers for cancer prognosis and possible therapeutic targets.
肿瘤浸润免疫细胞塑造肿瘤微环境,并与临床结局密切相关。一些转录因子(TFs)也被报道可以调节抗肿瘤活性和免疫细胞浸润。本研究旨在使用 CIBERSORT 和 EPIC 算法,根据 50 名癌症患者的批量 RNA 测序数据,定量分析肿瘤样本中不同免疫细胞的群体。加权基因共表达网络分析(WGCNA)确定了与肿瘤发生和 CD4+记忆 T 细胞、树突状细胞和巨噬细胞激活强烈相关的特征基因模块。TF 基因 、 、 、 和 是与免疫相关基因相关的特征基因模块的子网络中的核心基因。对癌症基因组图谱(TCGA)癌症数据的分析证实了这些发现,并进一步表明,这些潜在的 TF 基因调节免疫浸润的表达,以及它们调节的免疫相关基因,与多种癌症患者的生存有关。对具有 RNA-seq 数据的 24 对配对样本进行了外显子组测序。表达数量性状基因座(eQTL)分析表明,与非 TF 基因相比,TF 基因侧翼区域的突变明显更为频繁,这表明这些 TF 基因在调节免疫浸润中起驱动作用。总之,本研究提出了一种识别调节免疫浸润的基因的实用方法。这些基因可能是癌症预后的潜在生物标志物和可能的治疗靶点。