Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA.
Department of Biochemistry.
Blood Adv. 2021 Dec 14;5(23):4831-4841. doi: 10.1182/bloodadvances.2021004750.
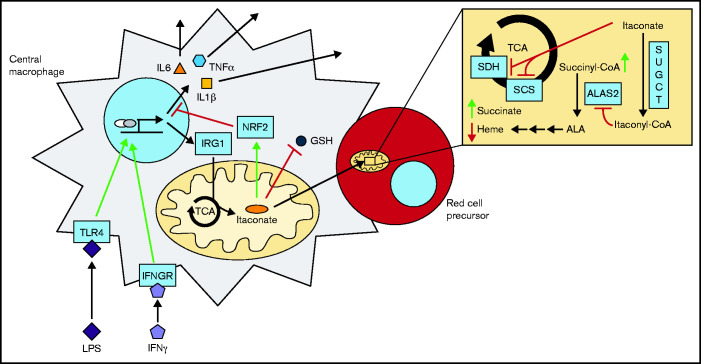
As part of the inflammatory response by macrophages, Irg1 is induced, resulting in millimolar quantities of itaconate being produced. This immunometabolite remodels the macrophage metabolome and acts as an antimicrobial agent when excreted. Itaconate is not synthesized within the erythron but instead may be acquired from central macrophages within the erythroid island. Previously, we reported that itaconate inhibits hemoglobinization of developing erythroid cells. Herein we show that this action is accomplished by inhibition of tetrapyrrole synthesis. In differentiating erythroid precursors, cellular heme and protoporphyrin IX synthesis are reduced by itaconate at an early step in the pathway. In addition, itaconate causes global alterations in cellular metabolite pools, resulting in elevated levels of succinate, 2-hydroxyglutarate, pyruvate, glyoxylate, and intermediates of glycolytic shunts. Itaconate taken up by the developing erythron can be converted to itaconyl-coenzyme A (CoA) by the enzyme succinyl-CoA:glutarate-CoA transferase. Propionyl-CoA, propionyl-carnitine, methylmalonic acid, heptadecanoic acid, and nonanoic acid, as well as the aliphatic amino acids threonine, valine, methionine, and isoleucine, are increased, likely due to the impact of endogenous itaconyl-CoA synthesis. We further show that itaconyl-CoA is a competitive inhibitor of the erythroid-specific 5-aminolevulinate synthase (ALAS2), the first and rate-limiting step in heme synthesis. These findings strongly support our hypothesis that the inhibition of heme synthesis observed in chronic inflammation is mediated not only by iron limitation but also by limitation of tetrapyrrole synthesis at the point of ALAS2 catalysis by itaconate. Thus, we propose that macrophage-derived itaconate promotes anemia during an inflammatory response in the erythroid compartment.
作为巨噬细胞炎症反应的一部分,Irg1 被诱导,导致产生毫摩尔数量的衣康酸。这种免疫代谢物重塑了巨噬细胞代谢组,并在排泄时作为一种抗菌剂发挥作用。衣康酸不是在红细胞内合成的,而是可能从红细胞岛内的中心巨噬细胞中获得。以前,我们报道过衣康酸抑制正在发育的红细胞的血红蛋白化。在此,我们表明这种作用是通过抑制四吡咯合成来实现的。在分化的红细胞前体中,衣康酸在途径的早期步骤中减少了细胞血红素和原卟啉 IX 的合成。此外,衣康酸导致细胞代谢物池的全局改变,导致琥珀酸、2-羟基戊二酸、丙酮酸、乙醛酸和糖酵解旁路的中间产物水平升高。进入正在发育的红细胞的衣康酸可以被酶琥珀酰-CoA:戊二酸-CoA 转移酶转化为衣康酰辅酶 A(CoA)。丙酰-CoA、丙酰肉碱、甲基丙二酸、十七烷酸和壬酸以及脂肪族氨基酸苏氨酸、缬氨酸、蛋氨酸和异亮氨酸也增加了,这可能是由于内源性衣康酰-CoA 合成的影响。我们进一步表明,衣康酰-CoA 是红细胞特异性 5-氨基酮戊酸合酶(ALAS2)的竞争性抑制剂,ALAS2 是血红素合成的第一步和限速步骤。这些发现强烈支持我们的假设,即慢性炎症中观察到的血红素合成抑制不仅是由铁限制引起的,而且还与衣康酸在 ALAS2 催化点限制四吡咯合成有关。因此,我们提出,巨噬细胞衍生的衣康酸在红细胞区的炎症反应中促进贫血。