Centre for Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, UK.
Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy.
J Neurol Neurosurg Psychiatry. 2022 Jan;93(1):48-56. doi: 10.1136/jnnp-2021-327186. Epub 2021 Sep 13.
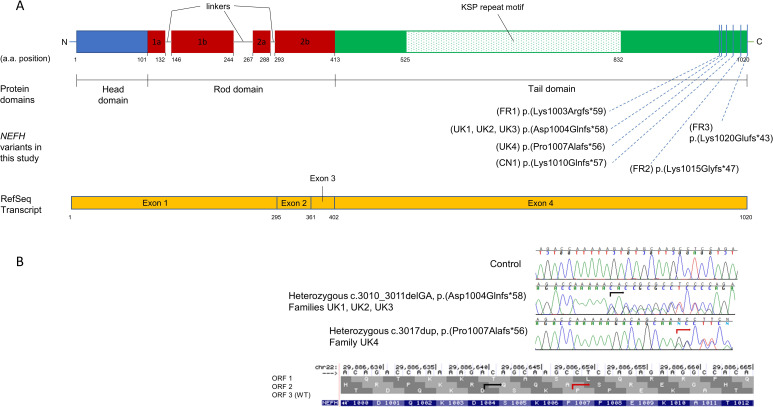
Neurofilaments are the major scaffolding proteins for the neuronal cytoskeleton, and variants in have recently been described to cause axonal Charcot-Marie-Tooth disease type 2CC (CMT2CC).
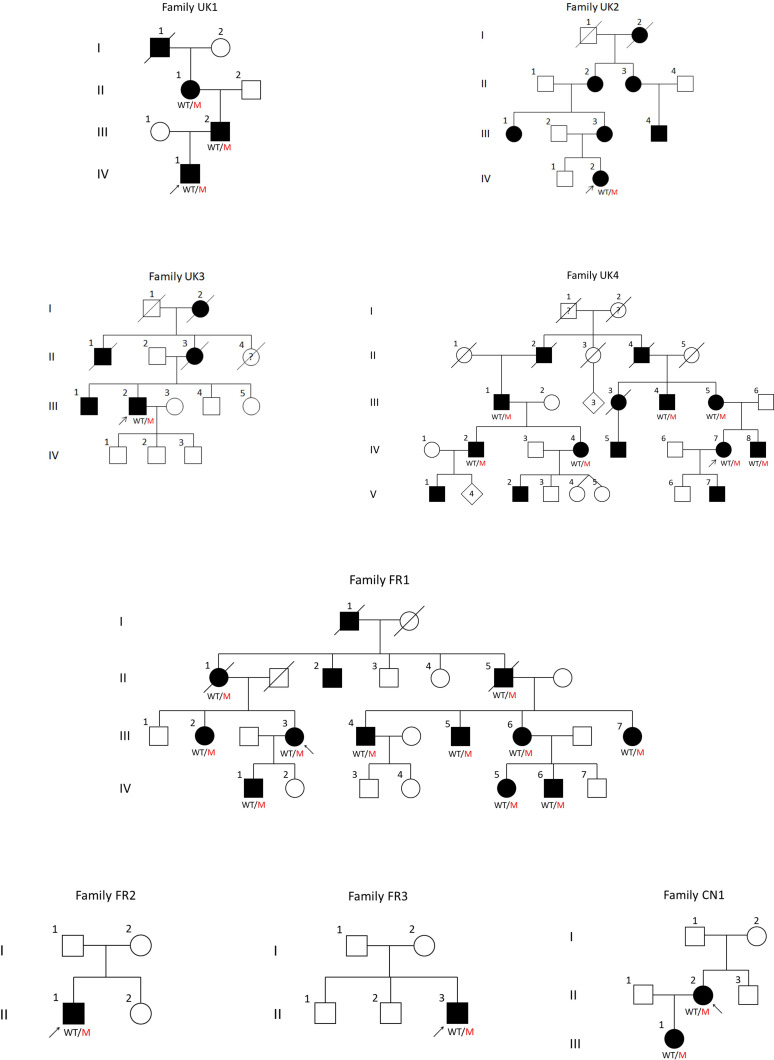
In this large observational study, we present phenotype-genotype correlations on 30 affected and 3 asymptomatic mutation carriers from eight families.
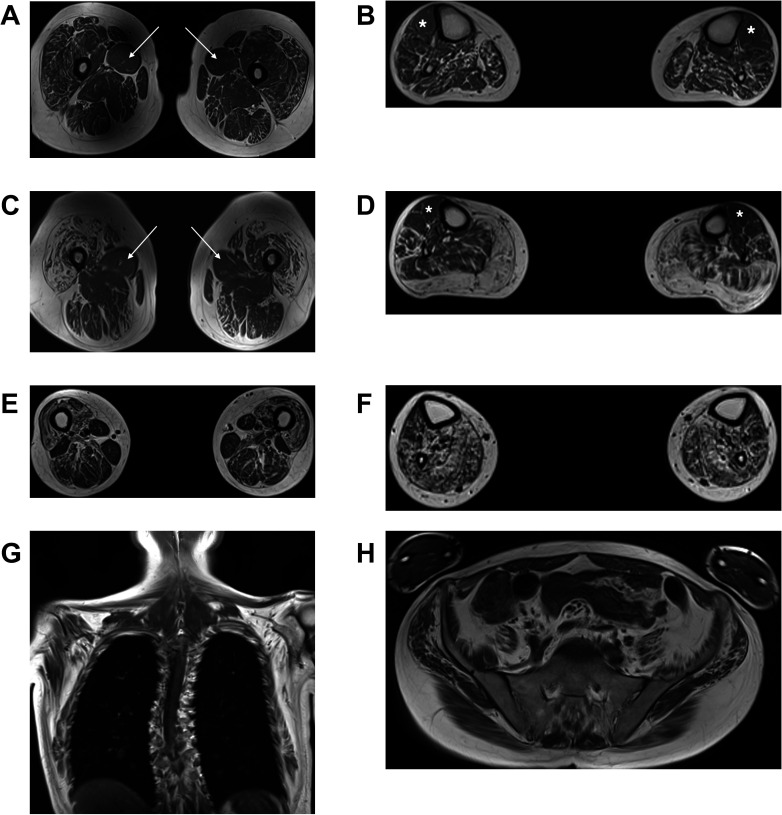
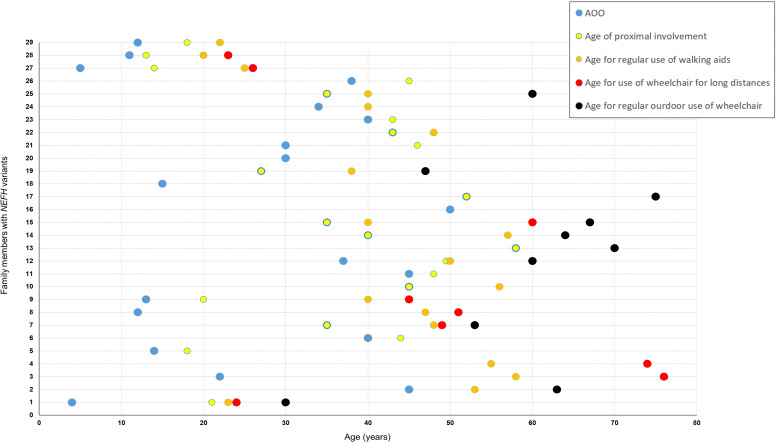
The majority of patients presented in adulthood with motor-predominant and lower limb-predominant symptoms and the average age of onset was 31.0±15.1 years. A prominent feature was the development of proximal weakness early in the course of the disease. The disease progressed rapidly, unlike other Charcot-Marie-Tooth disease (CMT) subtypes, and half of the patients (53%) needed to use a wheelchair on average 24.1 years after symptom onset. Furthermore, 40% of patients had evidence of early ankle plantarflexion weakness, a feature which is observed in only a handful of CMT subtypes. Neurophysiological studies and MRI of the lower limbs confirmed the presence of a non-length-dependent neuropathy in the majority of patients.All families harboured heterozygous frameshift variants in the last exon of , resulting in a reading frameshift to an alternate open reading frame and the translation of approximately 42 additional amino acids from the 3' untranslated region (3'-UTR).
This phenotype-genotype study highlights the unusual phenotype of CMT2CC, which is more akin to spinal muscular atrophy rather than classic CMT. Furthermore, the study will enable more informative discussions on the natural history of the disease and will aid in variant interpretation in the context of the disease's unique molecular genetics.
神经丝是神经元细胞骨架的主要支架蛋白,最近有研究表明 中的变异可导致轴索型 Charcot-Marie-Tooth 病 2CC(CMT2CC)。
在这项大型观察性研究中,我们对来自 8 个家系的 30 名受累者和 3 名无症状突变携带者进行了表型-基因型相关性分析。
大多数患者在成年期发病,表现为以运动为主、以下肢为主的症状,平均发病年龄为 31.0±15.1 岁。一个突出的特征是疾病早期就出现近端肌无力。与其他 Charcot-Marie-Tooth 病(CMT)亚型不同,疾病进展迅速,平均发病后 24.1 年,有一半的患者(53%)需要使用轮椅。此外,40%的患者出现早期踝关节跖屈肌无力的证据,这一特征仅见于少数几种 CMT 亚型。神经生理学研究和下肢 MRI 证实,大多数患者存在非长度依赖性神经病变。所有家系均携带 的最后一个外显子的杂合移码变异,导致阅读框移码,并从 3'非翻译区(3'-UTR)翻译出约 42 个额外的氨基酸。
本研究强调了 CMT2CC 的不典型表型,更类似于脊髓性肌萎缩症,而不是典型的 CMT。此外,该研究将能够更详细地讨论疾病的自然史,并有助于在疾病独特的分子遗传学背景下对 变异进行解释。