Ortore Rocco Pio, Leone Maria Pia, Palumbo Orazio, Petracca Antonio, Trecca Eleonora M C, D'Ecclesia Aurelio, Vigliaroli Ciro Lucio, Micale Lucia, Longo Francesco, Melchionda Salvatore, Castori Marco
Division of Maxillofacial Surgery and Otolaryngology, Fondazione IRCCS-Casa Sollievo della Sofferenza, San Giovanni Rotondo, 71013 Foggia, Italy.
Division of Medical Genetics, Fondazione IRCCS-Casa Sollievo della Sofferenza, San Giovanni Rotondo, 71013 Foggia, Italy.
Audiol Res. 2021 Sep 9;11(3):443-451. doi: 10.3390/audiolres11030041.
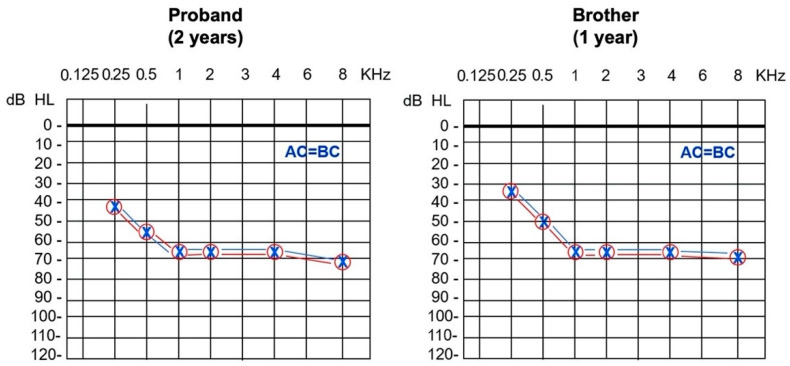
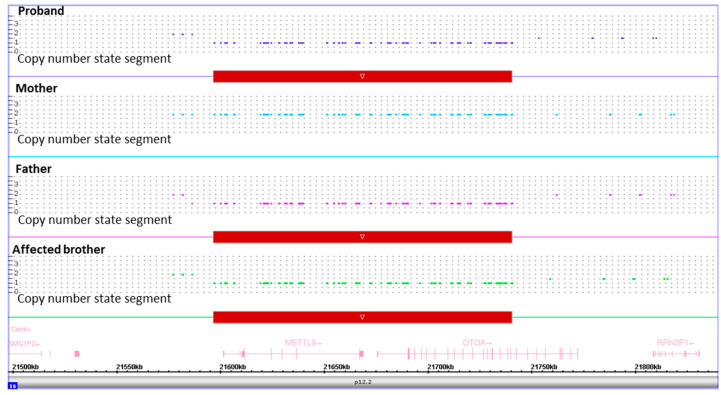
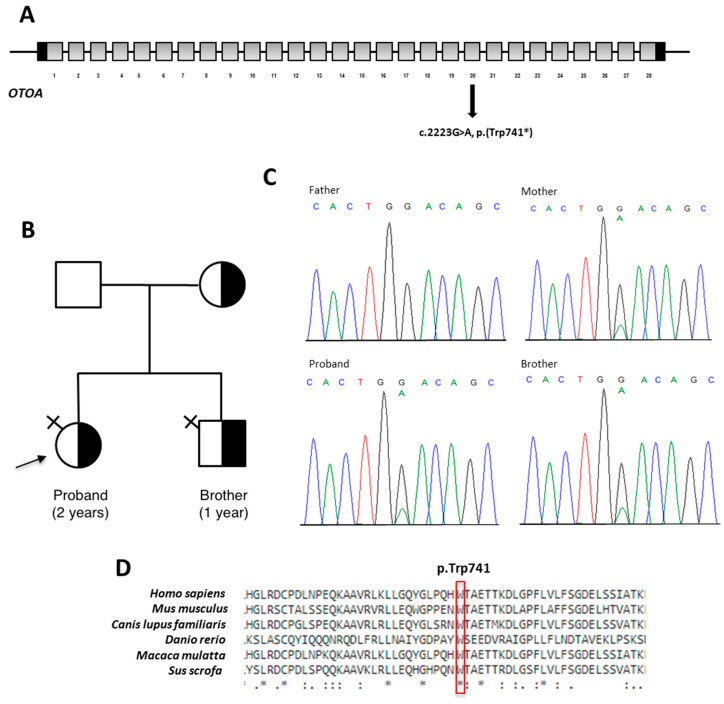
Hearing loss (HL) affects 1-3 newborns per 1000 and, in industrialized countries, recognizes a genetic etiology in more than 80% of the congenital cases. Excluding and , is one of the leading genes associated with autosomal recessive non-syndromic HL. Allelic heterogeneity linked to also includes genomic rearrangements facilitated by non-allelic homologous recombination with the neighboring pseudogene. We present a couple of Italian siblings affected by moderate to severe sensorineural hearing loss (SNHL) due to compound heterozygosity at the locus. Multigene panel next-generation sequencing identified the c.2223G>A, p.(Trp741*) variant transmitted from the unaffected mother. Assuming the existence of a second paternal deleterious variant which evaded detection at sequencing, genomic array analysis found a ~150 Kb microdeletion of paternal origin and spanning part of . Both deleterious alleles were identified for the first time. This study demonstrates the utility of an integrated approach to solve complex cases and allow appropriate management to affected individuals and at-risk relatives.
听力损失(HL)在每1000名新生儿中影响1至3名,在工业化国家,超过80%的先天性病例可归因于遗传病因。排除[具体基因1]和[具体基因2]后,[目标基因]是与常染色体隐性非综合征性HL相关的主要基因之一。与[目标基因]相关的等位基因异质性还包括由与相邻[假基因名称]的非等位基因同源重组促进的基因组重排。我们报告了一对意大利兄弟姐妹,他们因[目标基因]位点的复合杂合性而患有中度至重度感音神经性听力损失(SNHL)。多基因panel下一代测序确定了从未受影响的母亲遗传而来的c.2223G>A,p.(Trp741*)变异。假设存在第二个在测序时未被检测到的父系有害变异,基因组阵列分析发现了一个约150 Kb的父系来源微缺失,该微缺失跨越了[目标基因]的一部分。这两个有害等位基因均为首次鉴定。本研究证明了采用综合方法解决复杂病例并为受影响个体和高危亲属提供适当管理的实用性。