Institute of Evolutionary Biology, Faculty of Biology & Biological and Chemical Research Centre, University of Warsaw, Warszawa 02-089, Poland.
Bioinformatics. 2022 Jan 3;38(2):344-350. doi: 10.1093/bioinformatics/btab672.
With a large number of metagenomic datasets becoming available, eukaryotic metagenomics emerged as a new challenge. The proper classification of eukaryotic nuclear and organellar genomes is an essential step toward a better understanding of eukaryotic diversity.
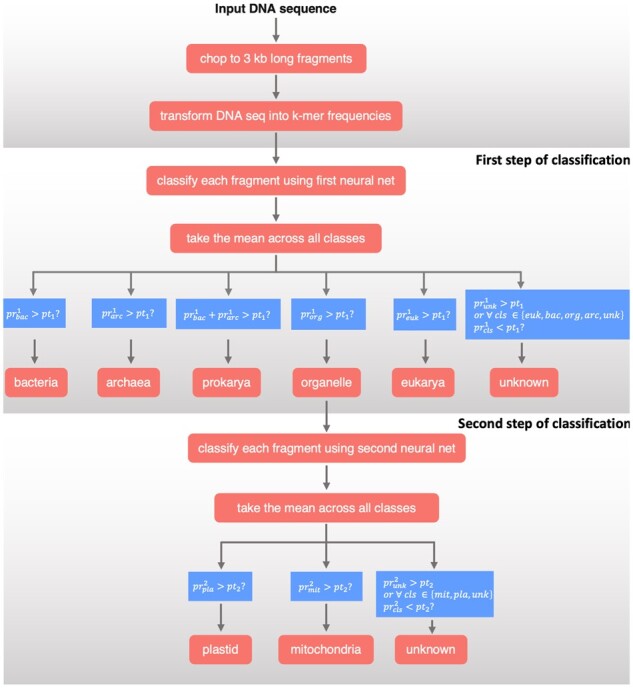
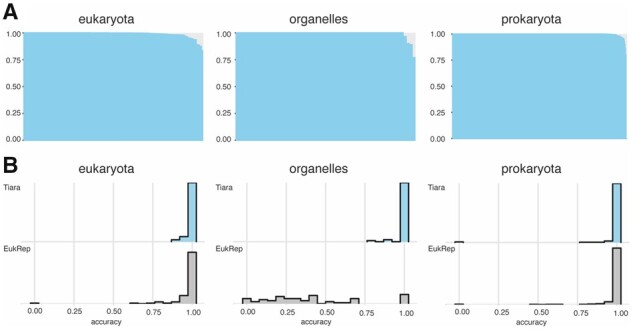
We developed Tiara, a deep-learning-based approach for the identification of eukaryotic sequences in the metagenomic datasets. Its two-step classification process enables the classification of nuclear and organellar eukaryotic fractions and subsequently divides organellar sequences into plastidial and mitochondrial. Using the test dataset, we have shown that Tiara performed similarly to EukRep for prokaryotes classification and outperformed it for eukaryotes classification with lower calculation time. In the tests on the real data, Tiara performed better than EukRep in analyzing the small dataset representing eukaryotic cell microbiome and large dataset from the pelagic zone of oceans. Tiara is also the only available tool correctly classifying organellar sequences, which was confirmed by the recovery of nearly complete plastid and mitochondrial genomes from the test data and real metagenomic data.
Tiara is implemented in python 3.8, available at https://github.com/ibe-uw/tiara and tested on Unix-based systems. It is released under an open-source MIT license and documentation is available at https://ibe-uw.github.io/tiara. Version 1.0.1 of Tiara has been used for all benchmarks.
Supplementary data are available at Bioinformatics online.
随着大量宏基因组数据集的出现,真核生物宏基因组学成为一个新的挑战。正确分类真核生物核和细胞器基因组是更好地理解真核生物多样性的关键步骤。
我们开发了 Tiara,这是一种基于深度学习的方法,用于鉴定宏基因组数据集中的真核序列。它的两步分类过程能够对核和细胞器真核部分进行分类,并随后将细胞器序列分为质体和线粒体。使用测试数据集,我们表明 Tiara 在原核生物分类方面的表现与 EukRep 相似,而在真核生物分类方面的表现优于 EukRep,且计算时间更短。在对真实数据的测试中,Tiara 在分析代表真核细胞微生物组的小数据集和来自海洋远洋区的大数据集方面的表现优于 EukRep。Tiara 也是唯一能够正确分类细胞器序列的可用工具,这一点通过从测试数据和真实宏基因组数据中恢复几乎完整的质体和线粒体基因组得到了证实。
Tiara 是用 python 3.8 编写的,可在 https://github.com/ibe-uw/tiara 上获得,并在基于 Unix 的系统上进行了测试。它是在开源 MIT 许可证下发布的,文档可在 https://ibe-uw.github.io/tiara 上获得。Tiara 的 1.0.1 版本已用于所有基准测试。
补充数据可在《生物信息学》在线获得。