Beijing Key Laboratory for Genetics of Birth Defects, Beijing Pediatric Research Institute, MOE Key Laboratory of Major Diseases in Children, Beijing Children's Hospital, Capital Medical University, National Center for Children's Health, Beijing, 100045, China.
Henan Key Laboratory of Pediatric Inherited and Metabolic Diseases, Henan Children's Hospital, Zhengzhou Hospital of Beijing Children's Hospital, Zhengzhou, China.
Orphanet J Rare Dis. 2021 Sep 29;16(1):403. doi: 10.1186/s13023-021-02028-4.
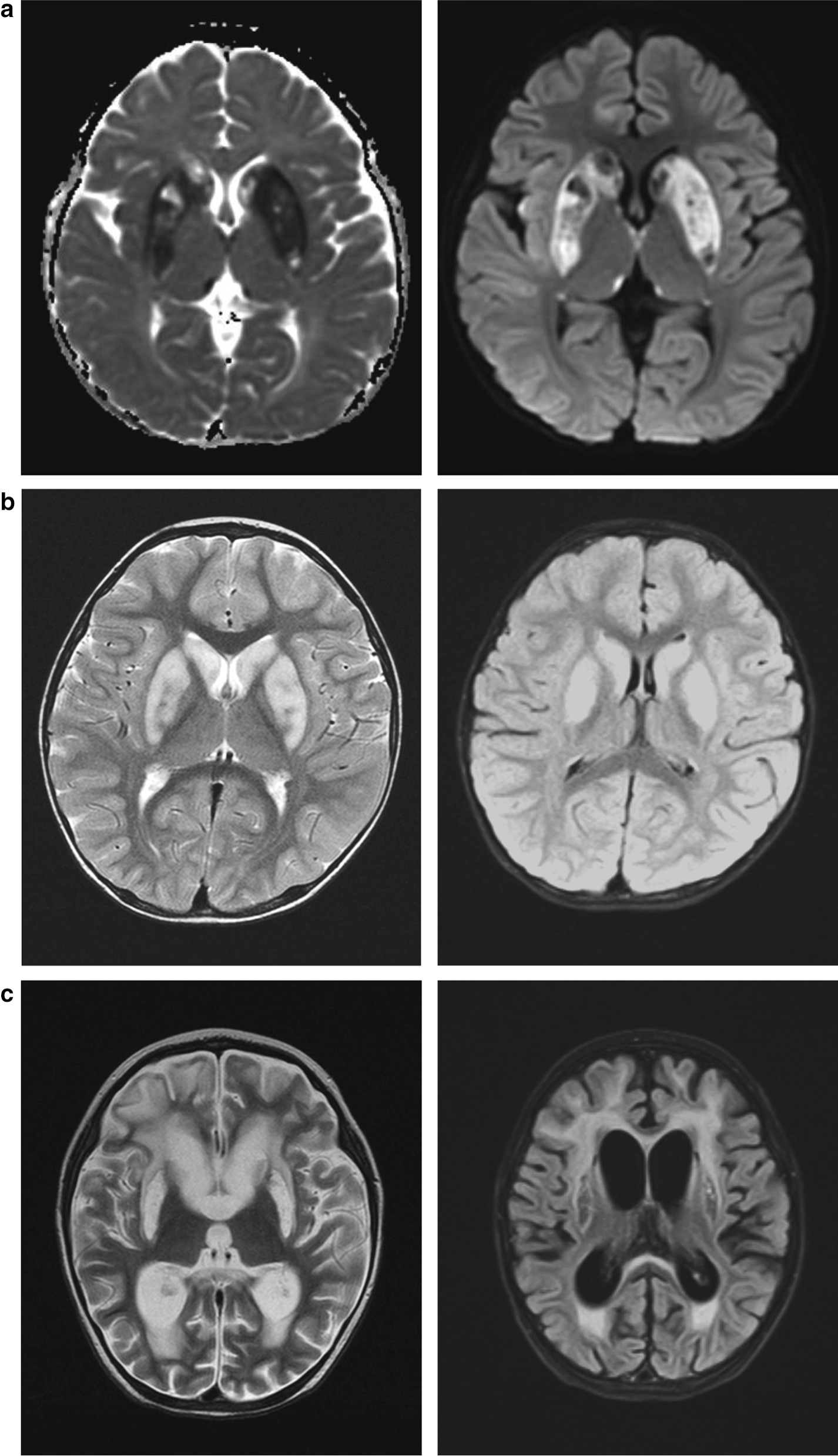
Thiamine metabolism dysfunction syndrome 4 (THMD4, OMIM #613710) is an autosomal recessive inherited disease caused by the deficiency of SLC25A19 that encodes the mitochondrial thiamine pyrophosphate (TPP) transporter. This disorder is characterized by bilateral striatal degradation and progressive polyneuropathy with the onset of fever of unknown origin. The limited number of reported cases and lack of functional annotation of related gene variants continue to limit diagnosis.
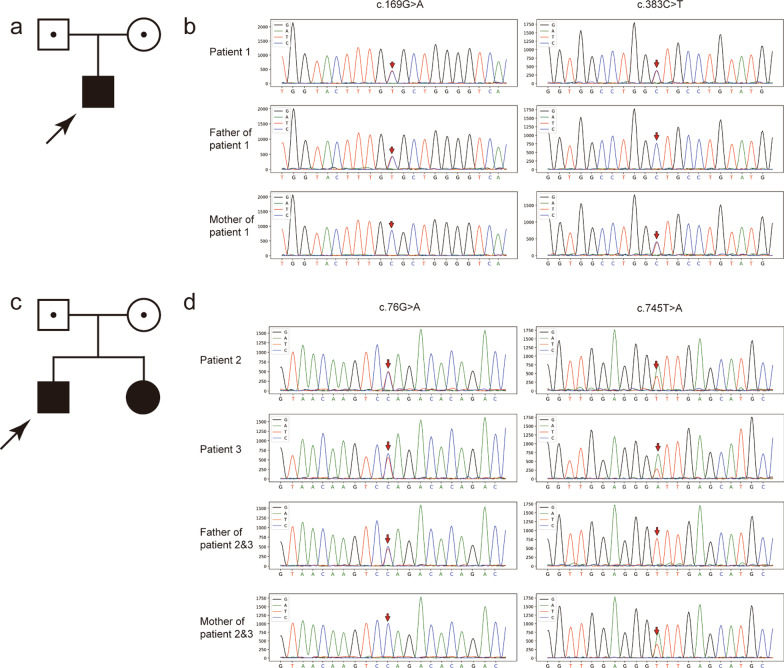
We report three cases of encephalopathy from two unrelated pedigrees with basal ganglia signal changes after fever of unknown origin. To distinguish this from other types of encephalopathy, such as acute necrotizing encephalopathy, exome sequencing was performed, and four novel heterozygous variations, namely, c.169G>A (p.Ala57Thr), c.383C>T (p.Ala128Val), c.76G>A (p.Gly26Arg), and c.745T>A (p.Phe249Ile), were identified in SLC25A19. All variants were confirmed using Sanger sequencing. To determine the pathogenicity of these variants, functional studies were performed. We found that mitochondrial TPP levels were significantly decreased in the presence of SLC25A19 variants, indicating that TPP transport activities of mutated SLC25A19 proteins were impaired. Thus, combining clinical phenotype, genetic analysis, and functional studies, these variants were deemed as likely pathogenic.
Exome sequencing analysis enables molecular diagnosis as well as provides potential etiology. Further studies will enable the elucidation of SLC25A19 protein function. Our investigation supplied key molecular evidence for the precise diagnosis of and clinical decision-making for a rare disease.
硫胺素代谢功能障碍综合征 4 型(THMD4,OMIM#613710)是一种常染色体隐性遗传疾病,由编码线粒体硫胺素焦磷酸(TPP)转运体的 SLC25A19 缺乏引起。这种疾病的特征是双侧纹状体退化和进行性多发性神经病,伴有不明原因的发热。由于报告的病例数量有限,以及相关基因变异的功能注释缺乏,诊断仍然受到限制。
我们报告了两例无关家系中 3 例发热后出现基底节信号改变的脑病病例。为了将其与其他类型的脑病(如急性坏死性脑病)区分开来,我们进行了外显子组测序,并在 SLC25A19 中发现了四个新的杂合变异,即 c.169G>A(p.Ala57Thr)、c.383C>T(p.Ala128Val)、c.76G>A(p.Gly26Arg)和 c.745T>A(p.Phe249Ile)。所有变异均通过 Sanger 测序进行了确认。为了确定这些变异的致病性,我们进行了功能研究。我们发现,存在 SLC25A19 变异时,线粒体 TPP 水平显著降低,表明突变的 SLC25A19 蛋白的 TPP 转运活性受损。因此,结合临床表型、遗传分析和功能研究,这些变异被认为可能是致病性的。
外显子组测序分析不仅可以进行分子诊断,还可以提供潜在的病因。进一步的研究将阐明 SLC25A19 蛋白的功能。我们的研究为阐明这种罕见疾病的精确诊断和临床决策提供了关键的分子证据。