Cancer Research and Molecular Biology Laboratories, Department of Biochemistry, University of Ibadan, Ibadan, Nigeria.
Cancer Research and Molecular Biology Laboratories, Department of Biochemistry, University of Ibadan, Ibadan, Nigeria..
Infect Genet Evol. 2021 Dec;96:105097. doi: 10.1016/j.meegid.2021.105097. Epub 2021 Oct 1.
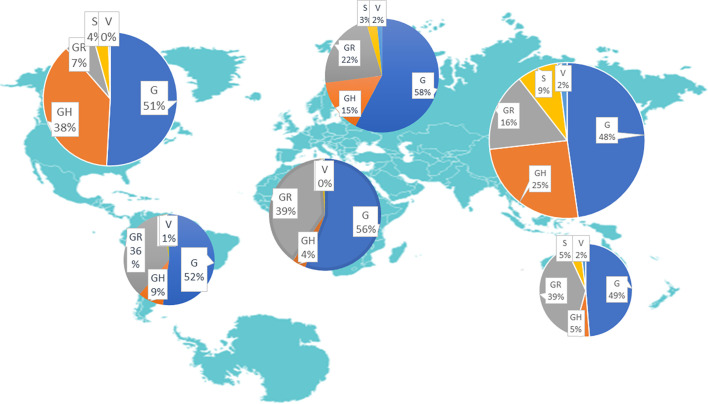
Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2 pathogen, has led to waves of global pandemic claiming lives and posing a serious threat to public health and social cum physical interactions. To evaluate the mutational landscape and conserved regions in the genome of the causative pathogen, we analysed 7213 complete SARS-CoV-2 protein sequences mined from the Global Initiative on Sharing All Influenza Data (GISAID) repository from infected patients across all regions on the EpiCov web interface. Regions of origin and the corresponding number of sequences mined are as follows: Asia - 2487; Oceania - 2027; Europe - 1240; Africa - 717; South America - 391; and North America - 351. High recurrent mutations, namely: T265I in non-structural protein 2 (nsp2), L3606F in nsp6, P4715L in RNA-dependent RNA polymerase (RdRp), D614G in spike glycoprotein, R203K and G204R in nucleocapsid phosphoprotein and Q57H in ORF3a with well-conserved envelope and membrane proteins, 3CLpro and spike S2 domains across regions were observed. Comparative analyses of the viral sequences reveal the prevalence P4715L and D614G mutations as the most recurrent and concurrent in Africa (97.20%), Europe (89.83%) and moderately in Asia (61.60%). Mutation rates are central to viral transmissibility, evolution and virulence, which help them to invade host immunity and develop drug resistance. Based on the foregoing, it is important to understand the mutational spectra of SARS-CoV-2 genome across regions. This will help in identifying specific genomic sites as potential targets for drug design and vaccine development, monitoring the spread of the virus and unraveling its evolution, virulence and transmissibility.
新型冠状病毒病(COVID-19)是由 SARS-CoV-2 病原体引起的,它导致了一波又一波的全球大流行,夺走了生命,对公共卫生和社会及身体互动构成了严重威胁。为了评估病原体基因组中的突变景观和保守区域,我们在 EpiCov 网络界面上分析了从全球共享流感数据倡议(GISAID)存储库中感染患者中挖掘出的 7213 个完整的 SARS-CoV-2 蛋白质序列。起源地区和挖掘出的相应序列数量如下:亚洲 - 2487;大洋洲 - 2027;欧洲 - 1240;非洲 - 717;南美洲 - 391;北美洲 - 351。高复发突变,即:非结构蛋白 2(nsp2)中的 T265I、nsp6 中的 L3606F、RNA 依赖性 RNA 聚合酶(RdRp)中的 P4715L、刺突糖蛋白中的 D614G、核衣壳磷蛋白中的 R203K 和 G204R 以及 ORF3a 中的 Q57H,其包膜和膜蛋白、3CLpro 和刺突 S2 结构域在各区域都有很好的保守性。对病毒序列的比较分析显示,P4715L 和 D614G 突变在非洲(97.20%)、欧洲(89.83%)中非常普遍,在亚洲也较为常见(61.60%)。突变率是病毒传播性、进化和毒力的关键,这有助于它们入侵宿主免疫并产生耐药性。基于上述情况,了解 SARS-CoV-2 基因组在各区域的突变谱非常重要。这有助于确定特定的基因组位点作为药物设计和疫苗开发的潜在靶标,监测病毒的传播并揭示其进化、毒力和传播性。