UCL Genetics Institute, University College London, London WC1E 6BT, UK.
UCL Genetics Institute, University College London, London WC1E 6BT, UK.
Infect Genet Evol. 2020 Sep;83:104351. doi: 10.1016/j.meegid.2020.104351. Epub 2020 May 5.
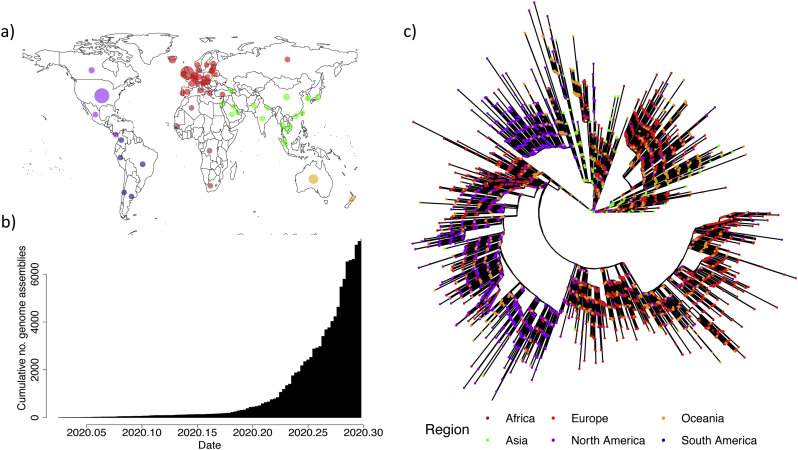
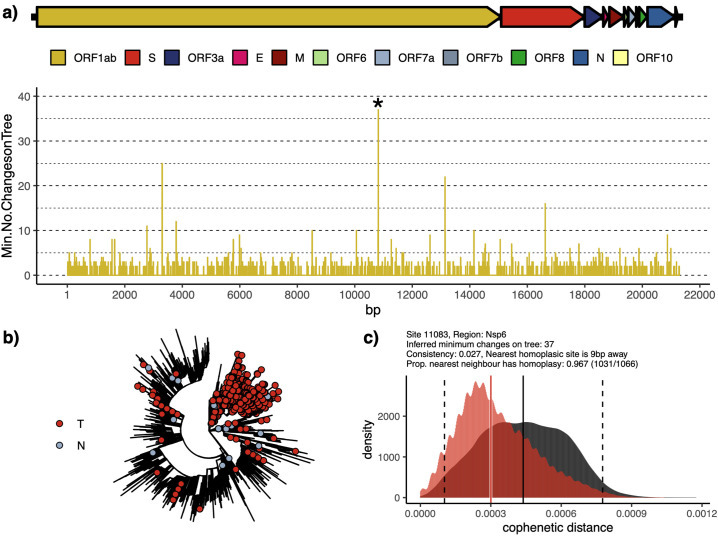
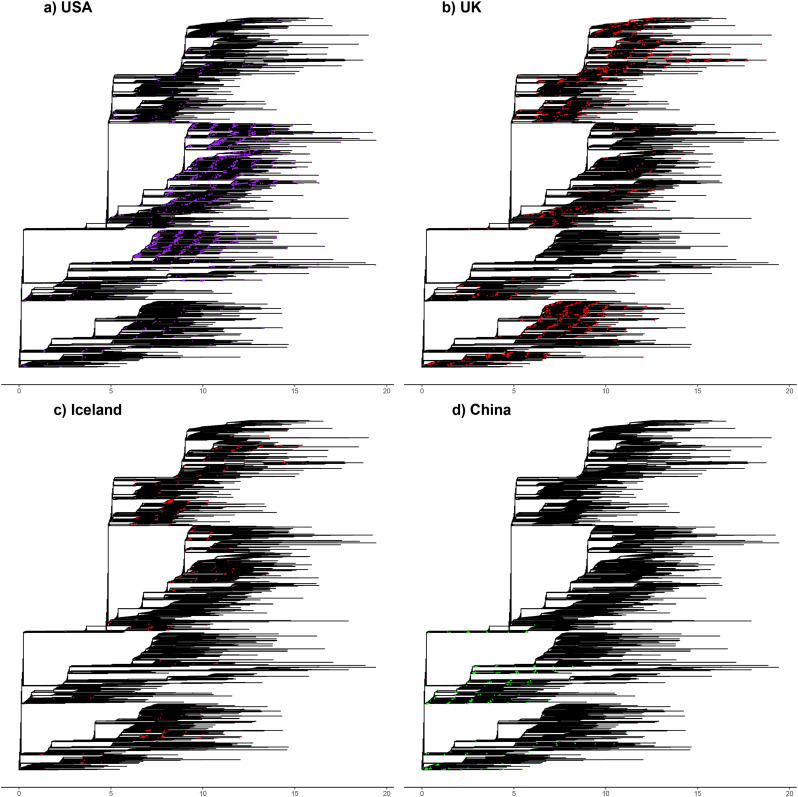
SARS-CoV-2 is a SARS-like coronavirus of likely zoonotic origin first identified in December 2019 in Wuhan, the capital of China's Hubei province. The virus has since spread globally, resulting in the currently ongoing COVID-19 pandemic. The first whole genome sequence was published on January 5 2020, and thousands of genomes have been sequenced since this date. This resource allows unprecedented insights into the past demography of SARS-CoV-2 but also monitoring of how the virus is adapting to its novel human host, providing information to direct drug and vaccine design. We curated a dataset of 7666 public genome assemblies and analysed the emergence of genomic diversity over time. Our results are in line with previous estimates and point to all sequences sharing a common ancestor towards the end of 2019, supporting this as the period when SARS-CoV-2 jumped into its human host. Due to extensive transmission, the genetic diversity of the virus in several countries recapitulates a large fraction of its worldwide genetic diversity. We identify regions of the SARS-CoV-2 genome that have remained largely invariant to date, and others that have already accumulated diversity. By focusing on mutations which have emerged independently multiple times (homoplasies), we identify 198 filtered recurrent mutations in the SARS-CoV-2 genome. Nearly 80% of the recurrent mutations produced non-synonymous changes at the protein level, suggesting possible ongoing adaptation of SARS-CoV-2. Three sites in Orf1ab in the regions encoding Nsp6, Nsp11, Nsp13, and one in the Spike protein are characterised by a particularly large number of recurrent mutations (>15 events) which may signpost convergent evolution and are of particular interest in the context of adaptation of SARS-CoV-2 to the human host. We additionally provide an interactive user-friendly web-application to query the alignment of the 7666 SARS-CoV-2 genomes.
SARS-CoV-2 是一种类似于 SARS 的冠状病毒,可能源自动物,于 2019 年 12 月在中国湖北省省会武汉首次被发现。该病毒随后在全球范围内传播,导致目前正在发生的 COVID-19 大流行。第一个完整的基因组序列于 2020 年 1 月 5 日公布,此后已对数千个基因组进行了测序。这一资源使我们能够以前所未有的方式了解 SARS-CoV-2 的过去种群动态,还能监测病毒如何适应其新的人类宿主,为指导药物和疫苗设计提供信息。我们整理了一个包含 7666 个公共基因组组装的数据集,并分析了随着时间的推移基因组多样性的出现。我们的结果与之前的估计一致,表明所有序列都有一个共同的祖先,这支持了 SARS-CoV-2 在 2019 年底进入人类宿主的时期。由于广泛传播,该病毒在几个国家的遗传多样性反映了其全球遗传多样性的很大一部分。我们确定了迄今为止 SARS-CoV-2 基因组中仍然基本不变的区域,以及已经积累了多样性的区域。通过关注已经独立多次出现的突变(同质性),我们在 SARS-CoV-2 基因组中鉴定了 198 个过滤后的反复出现的突变。在蛋白质水平上,近 80%的反复出现的突变产生了非同义变化,表明 SARS-CoV-2 可能正在适应。编码 Nsp6、Nsp11、Nsp13 的 Orf1ab 区域中的三个位点和 Spike 蛋白中的一个位点,其特征是反复出现的突变(>15 次)数量特别多,这可能标志着趋同进化,并且在 SARS-CoV-2 适应人类宿主的背景下特别有趣。我们还提供了一个交互式用户友好的网络应用程序,用于查询 7666 个 SARS-CoV-2 基因组的比对。