Department of Biological Chemistry & Pharmacology, The Ohio State University Wexner Medical Center, Columbus, Ohio, USA.
Department of Biochemistry & Molecular Biology, Holling Cancer Center, Medical University of South Carolina, Charleston, South Carolina, USA.
JCI Insight. 2021 Oct 8;6(19):e149023. doi: 10.1172/jci.insight.149023.
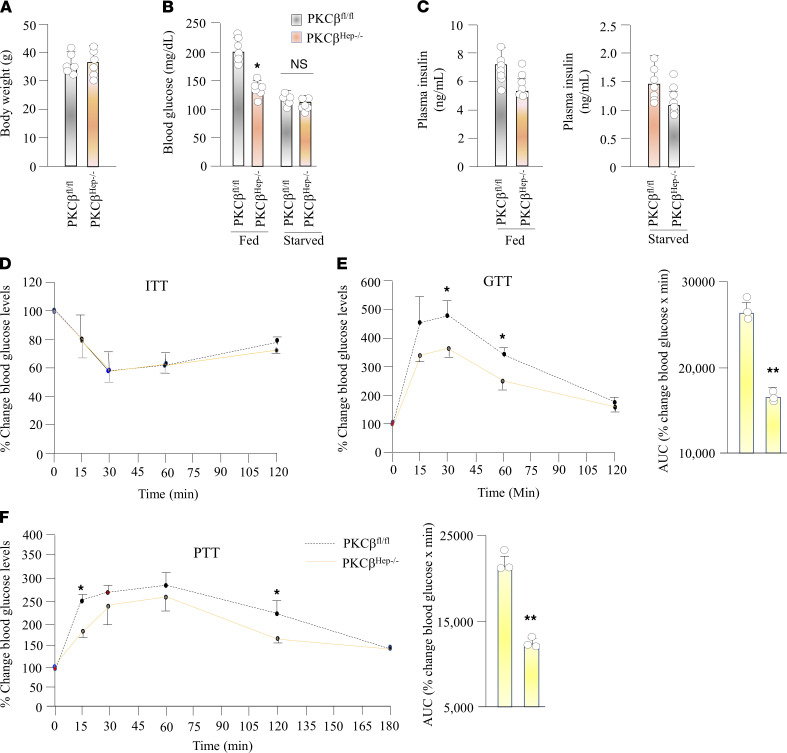
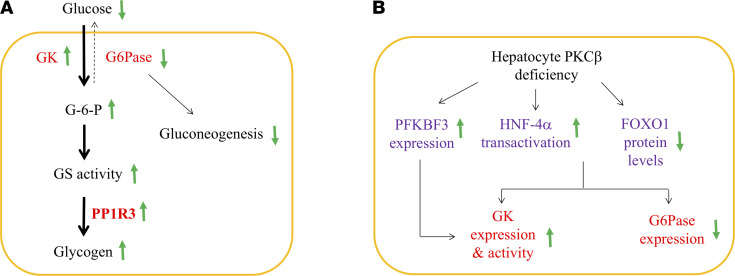
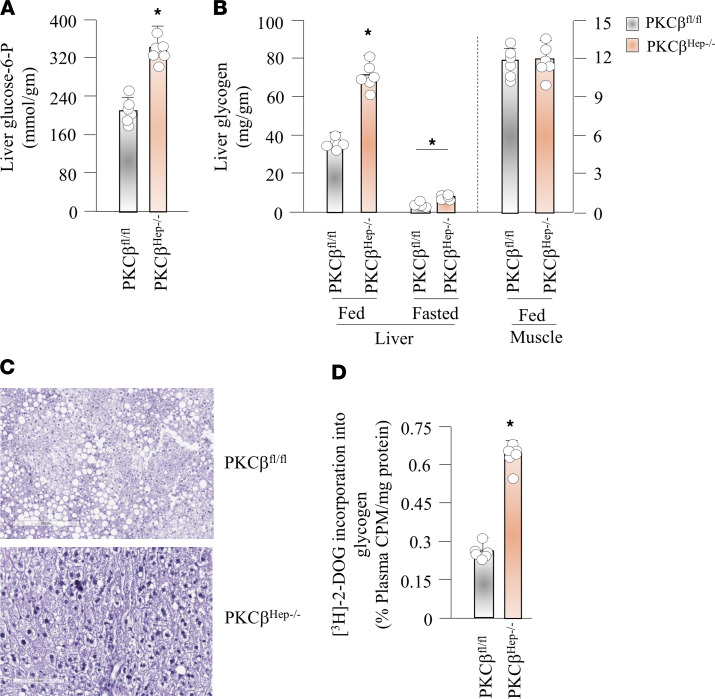
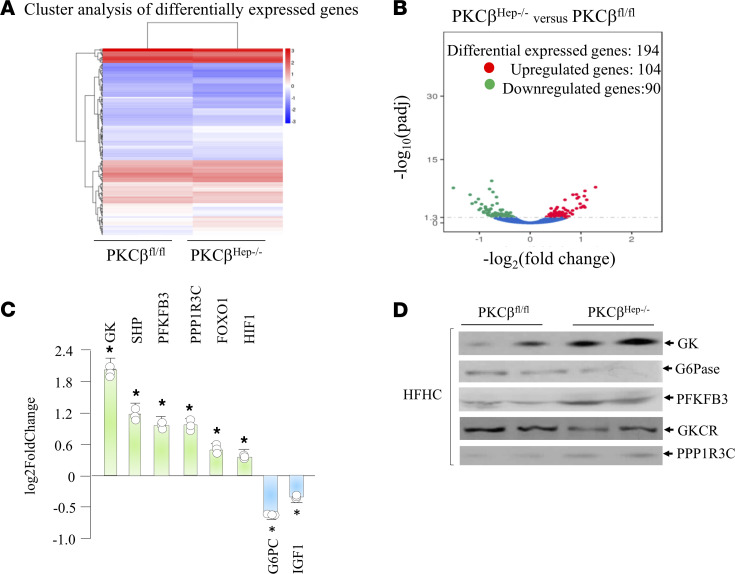
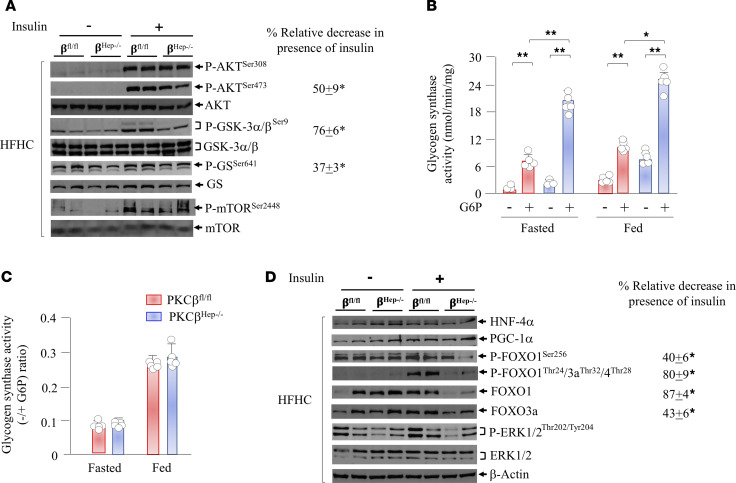
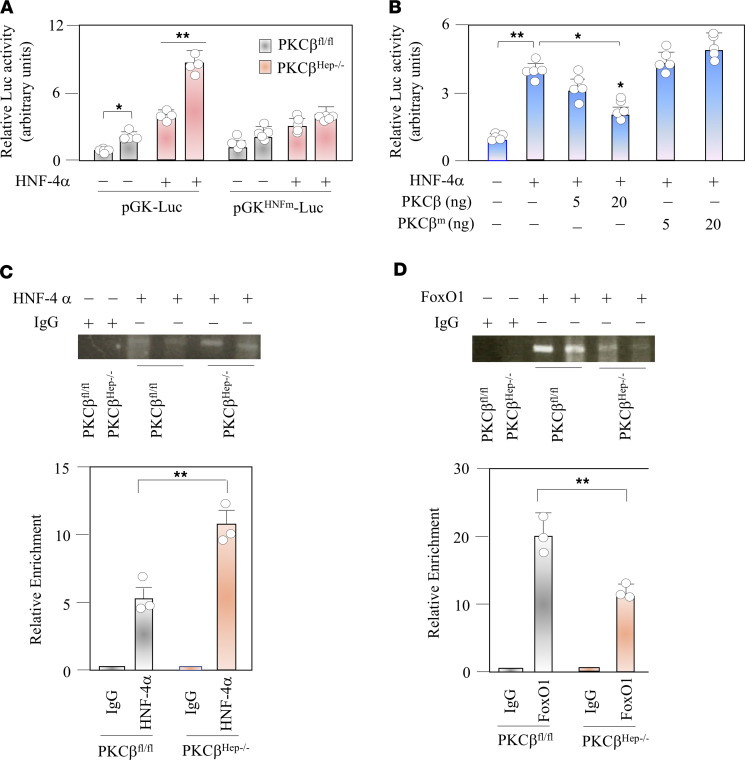
The signaling mechanisms by which dietary fat and cholesterol signals regulate central pathways of glucose homeostasis are not completely understood. By using a hepatocyte-specific PKCβ-deficient (PKCβHep-/-) mouse model, we demonstrated the role of hepatic PKCβ in slowing disposal of glucose overload by suppressing glycogenesis and increasing hepatic glucose output. PKCβHep-/- mice exhibited lower plasma glucose under the fed condition, modestly improved systemic glucose tolerance and mildly suppressed gluconeogenesis, increased hepatic glycogen accumulation and synthesis due to elevated glucokinase expression and activated glycogen synthase (GS), and suppressed glucose-6-phosphatase expression compared with controls. These events were independent of hepatic AKT/GSK-3α/β signaling and were accompanied by increased HNF-4α transactivation, reduced FoxO1 protein abundance, and elevated expression of GS targeting protein phosphatase 1 regulatory subunit 3C in the PKCβHep-/- liver compared with controls. The above data strongly imply that hepatic PKCβ deficiency causes hypoglycemia postprandially by promoting glucose phosphorylation via upregulating glucokinase and subsequently redirecting more glucose-6-phosphate to glycogen via activating GS. In summary, hepatic PKCβ has a unique and essential ability to induce a coordinated response that negatively affects glycogenesis at multiple levels under physiological postprandial conditions, thereby integrating nutritional fat intake with dysregulation of glucose homeostasis.
尚不完全清楚膳食脂肪和胆固醇信号调节葡萄糖稳态中枢途径的信号机制。通过使用肝特异性 PKCβ 缺陷(PKCβHep-/-)小鼠模型,我们证明了肝 PKCβ 在抑制糖生成和增加肝葡萄糖输出方面通过抑制葡萄糖过载的处置来发挥作用。与对照组相比,PKCβHep-/- 小鼠在进食状态下表现出较低的血浆葡萄糖水平,适度改善了全身葡萄糖耐量,轻度抑制了糖异生,由于升高的葡糖激酶表达和激活的糖原合酶(GS),肝糖原积累和合成增加,并且葡萄糖-6-磷酸酶表达受到抑制。这些事件独立于肝 AKT/GSK-3α/β 信号通路,并且伴随着 HNF-4α 反式激活增加、FoxO1 蛋白丰度降低以及在 PKCβHep-/- 肝中 GS 靶向蛋白磷酸酶 1 调节亚基 3C 的表达升高。上述数据强烈表明,肝 PKCβ 通过上调葡糖激酶促进葡萄糖磷酸化,从而导致餐后低血糖,随后通过激活 GS 将更多的葡萄糖-6-磷酸重新导向糖原。总之,肝 PKCβ 具有独特且必需的能力,可在生理餐后条件下通过多个水平负向影响糖生成来诱导协调反应,从而将营养脂肪摄入与葡萄糖稳态失调整合在一起。