Institute of Systems Biomedicine, Department of Medical Bioinformatics, School of Basic Medical Sciences, Peking University Health Science Center, Key Laboratory for Neuroscience, Ministry of Education/National Health Commission of China , NHC Key Laboratory of Medical Immunology (Peking University), Beijing, China.
Department of Human Anatomy, Histology & Embryology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China.
Elife. 2021 Oct 14;10:e69457. doi: 10.7554/eLife.69457.
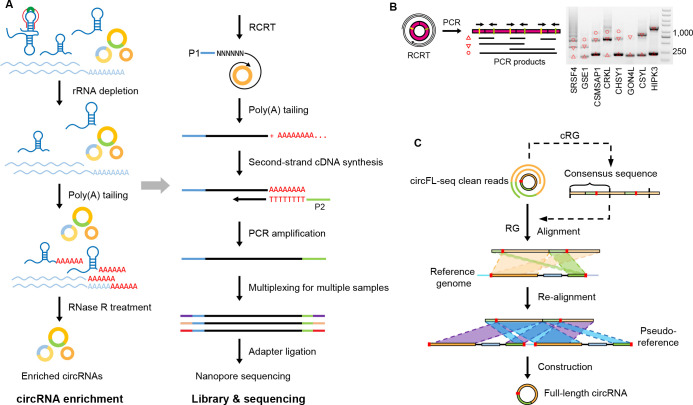
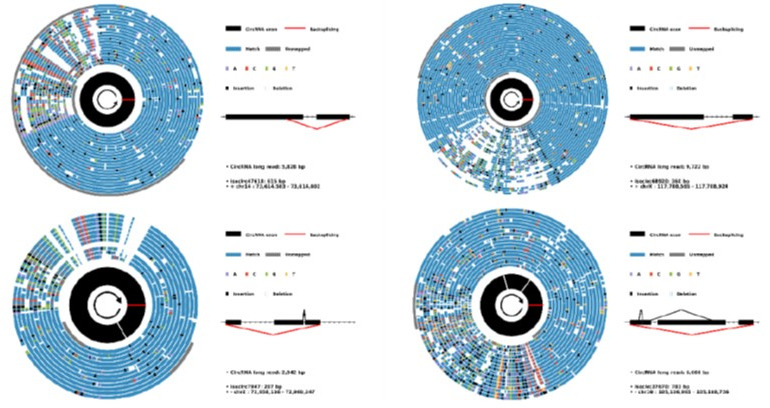
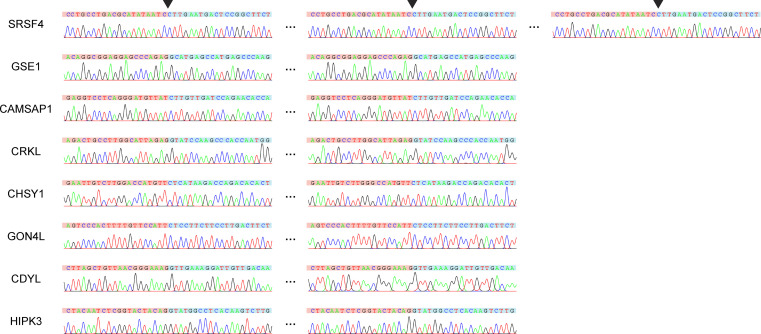
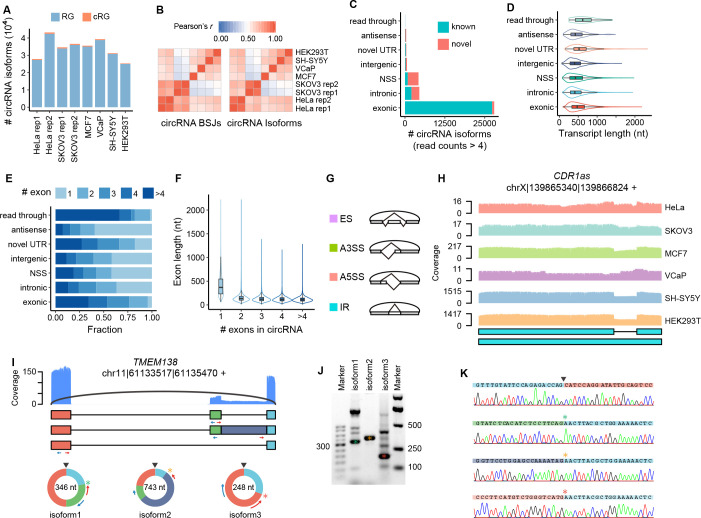
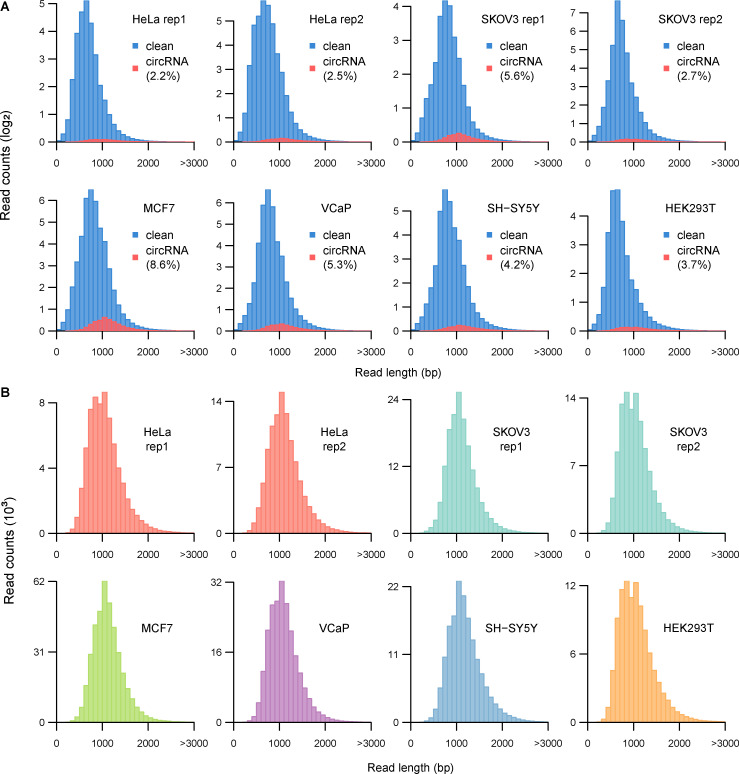
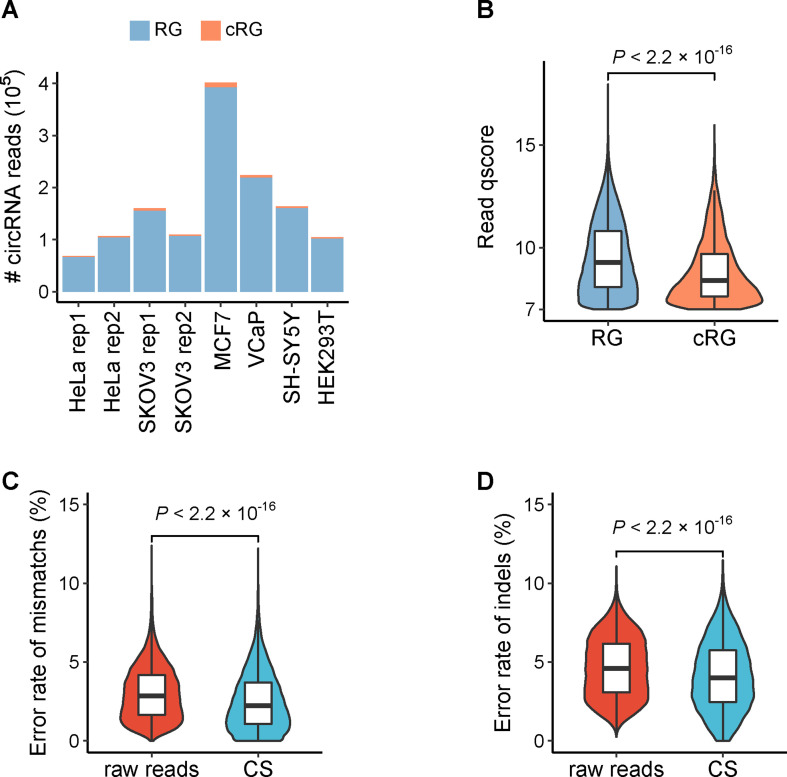
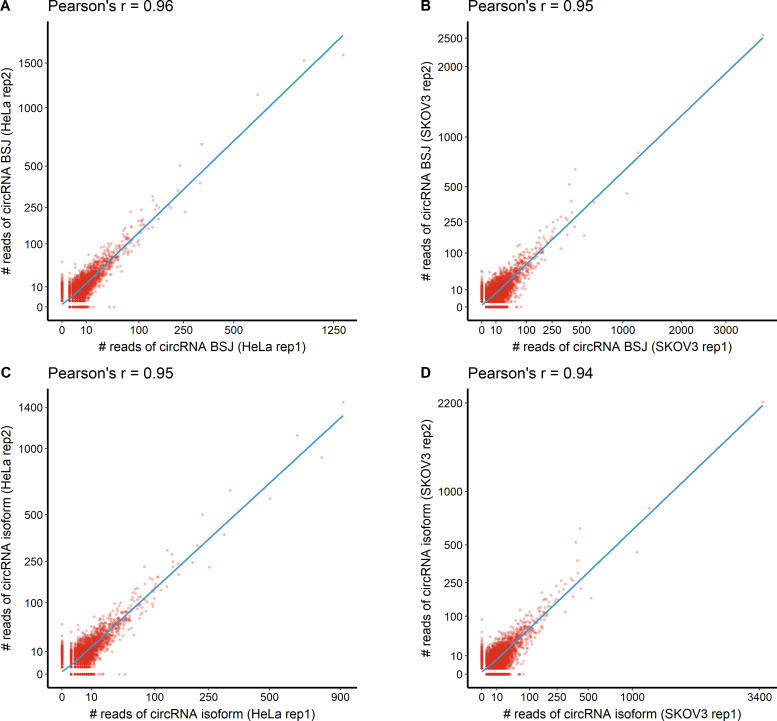
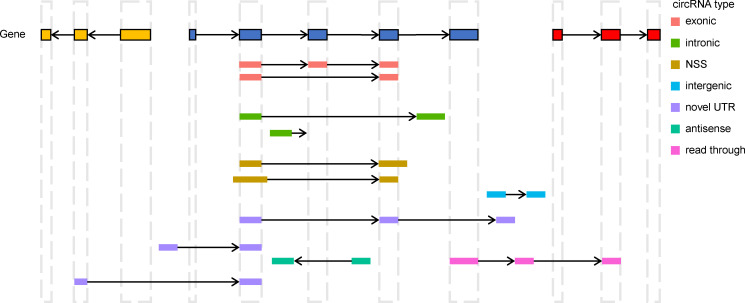
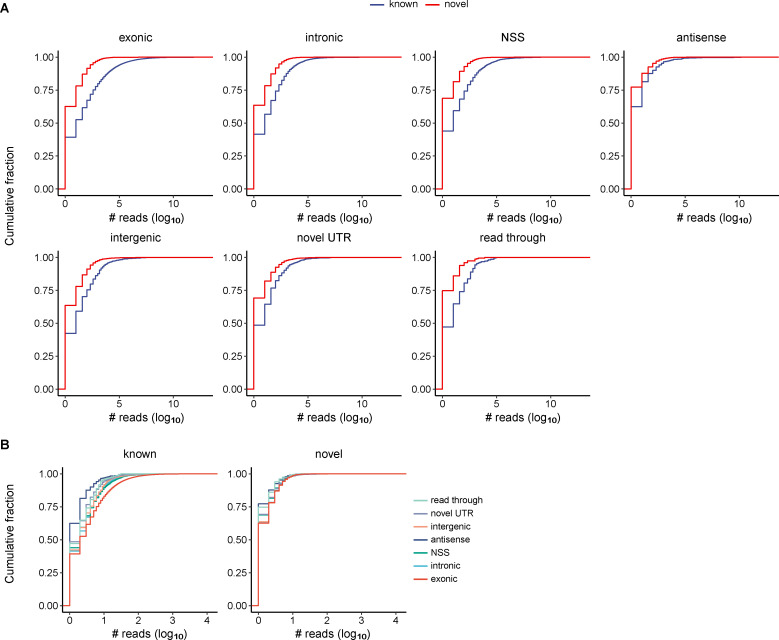
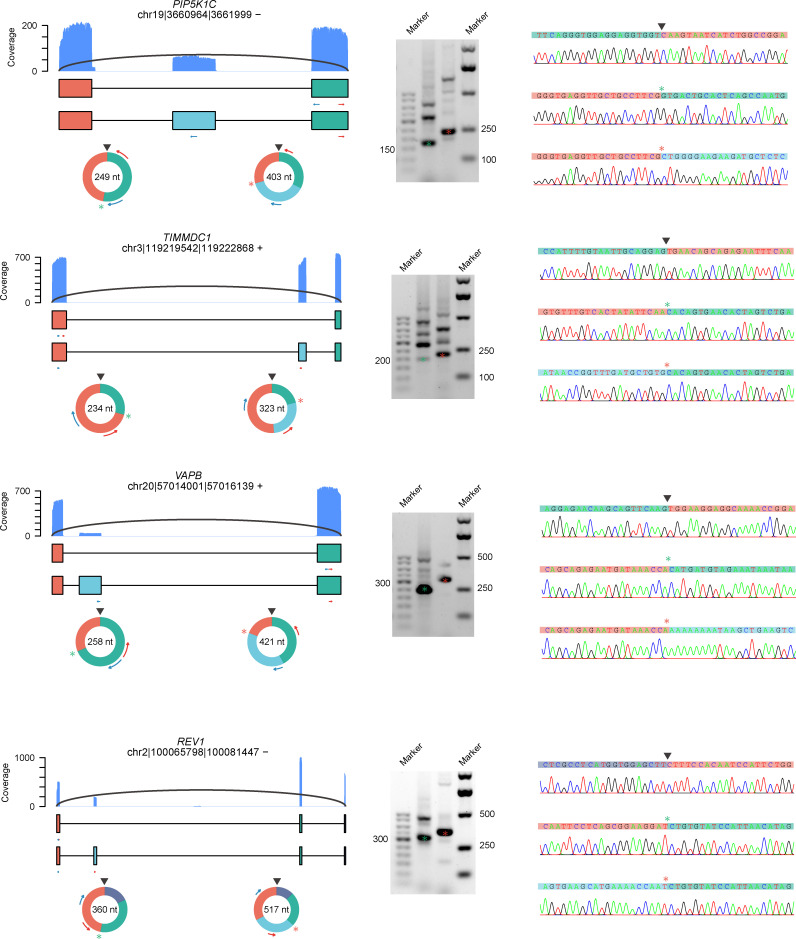
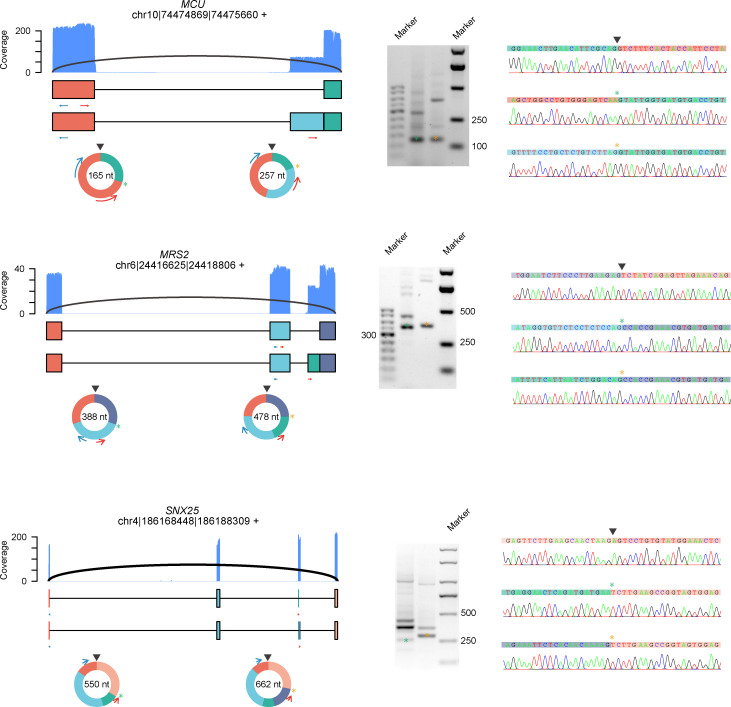
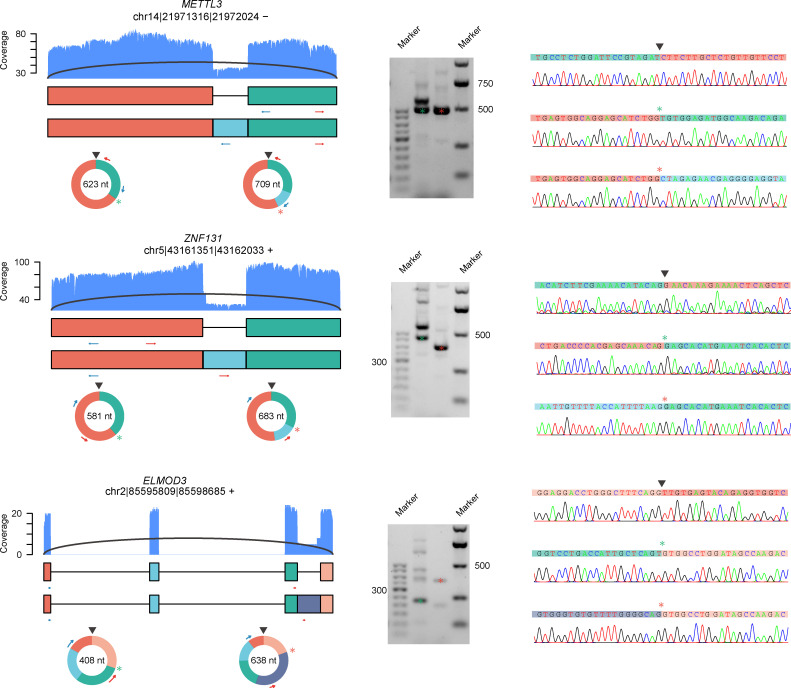
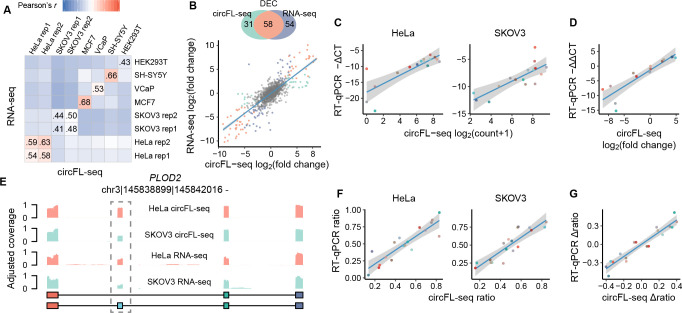
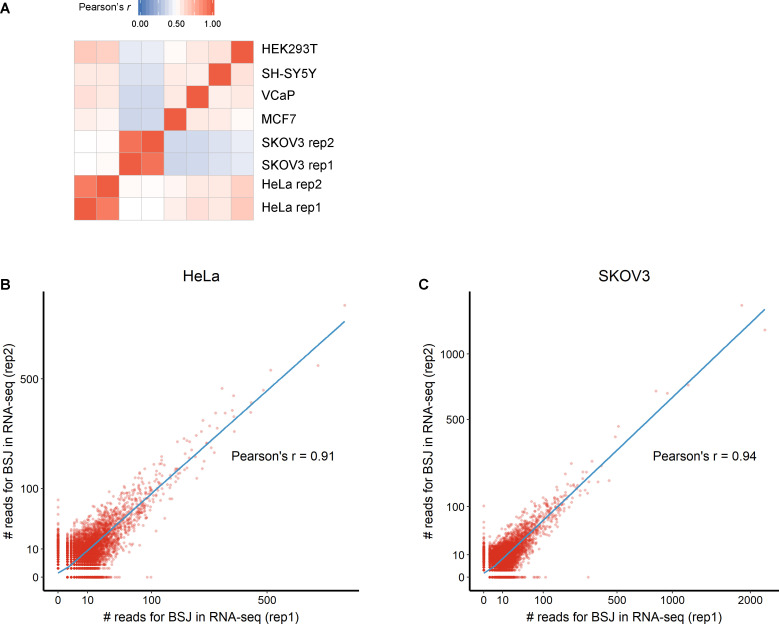
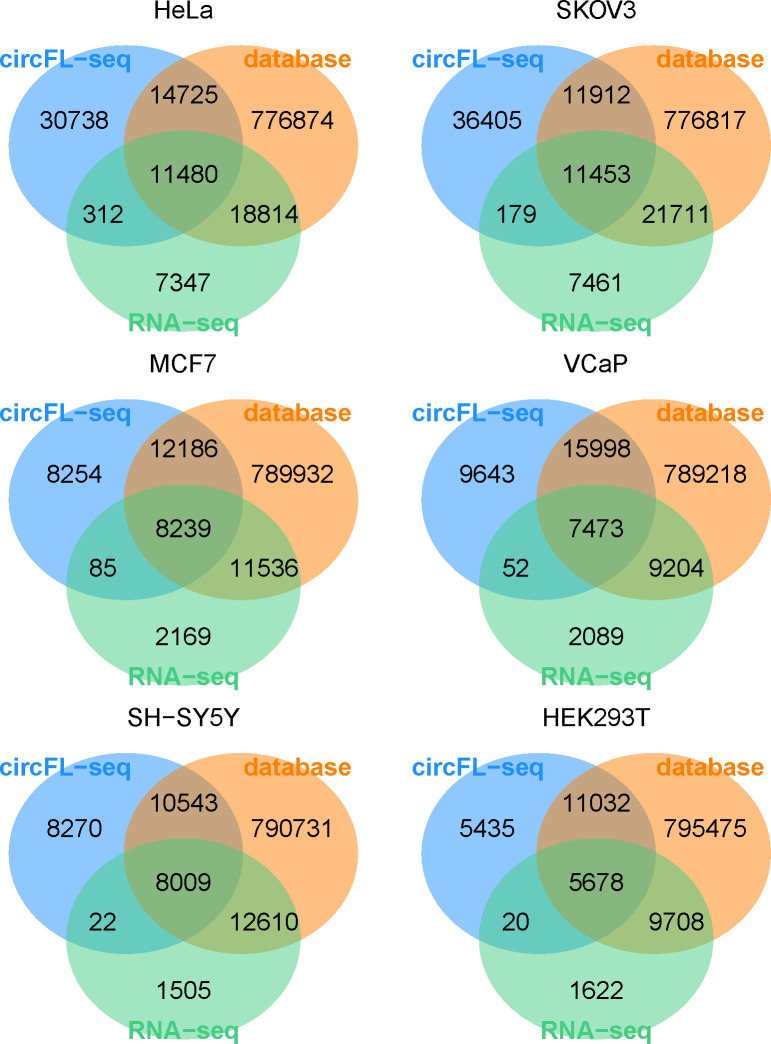
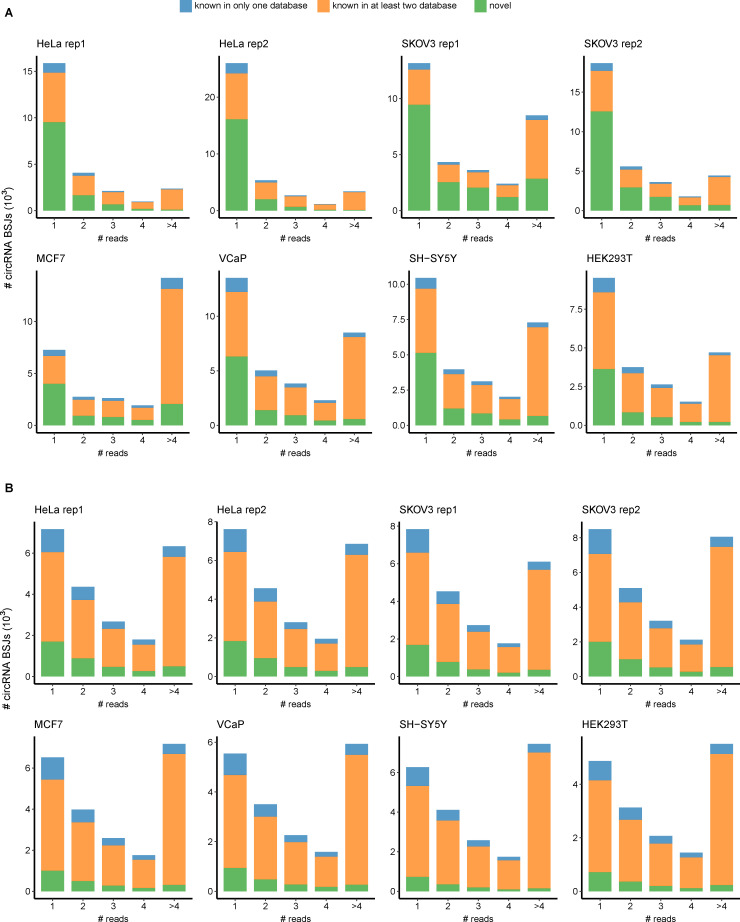
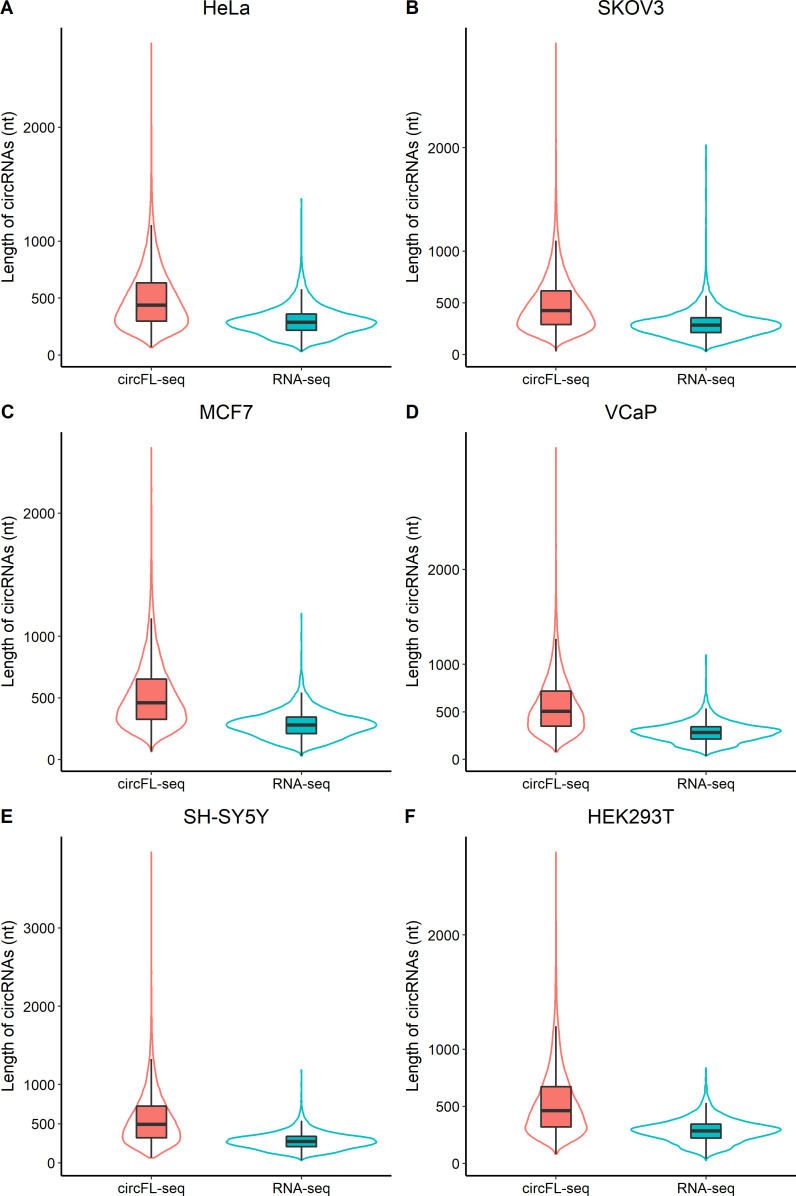
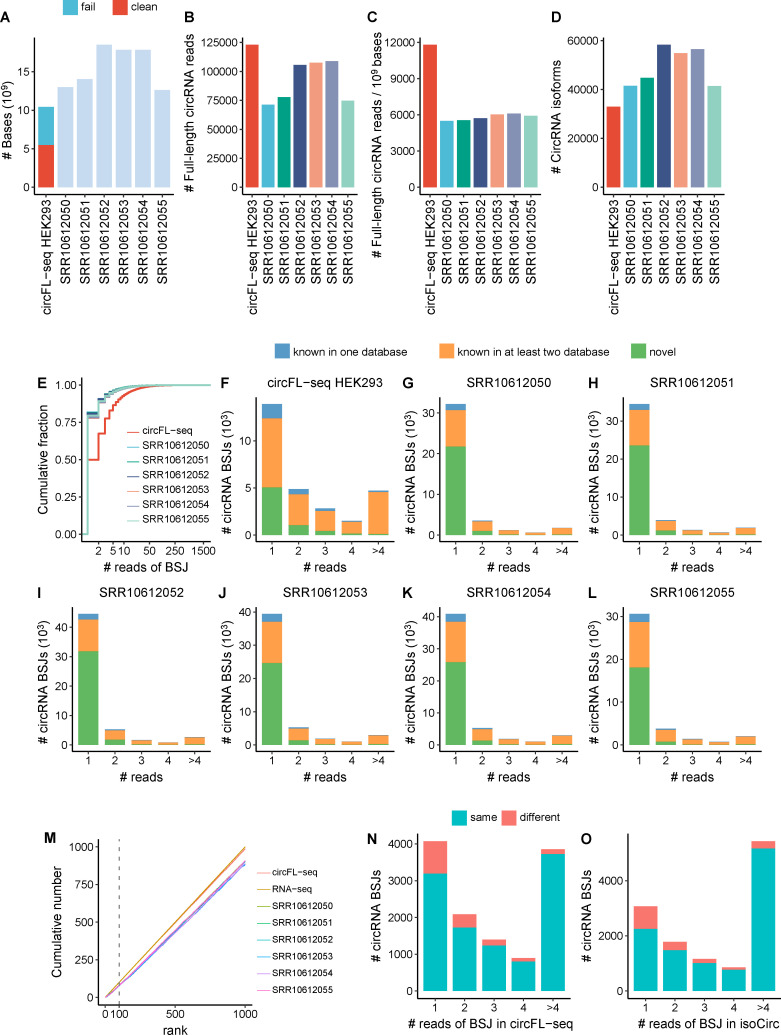
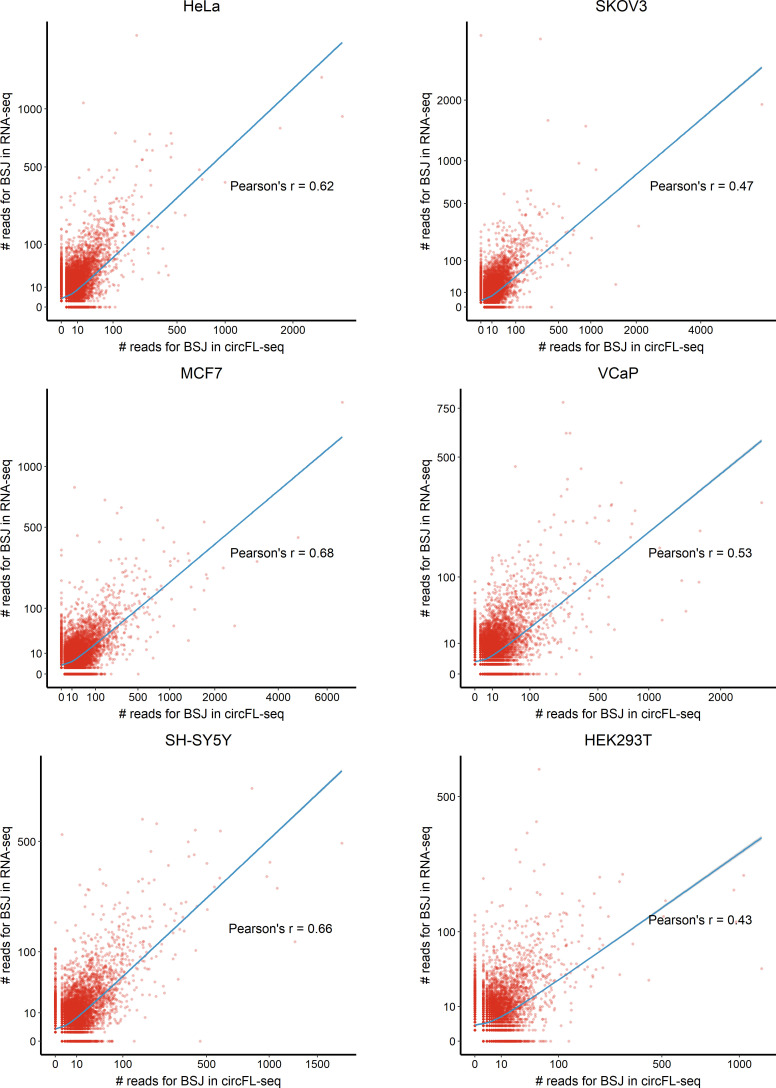
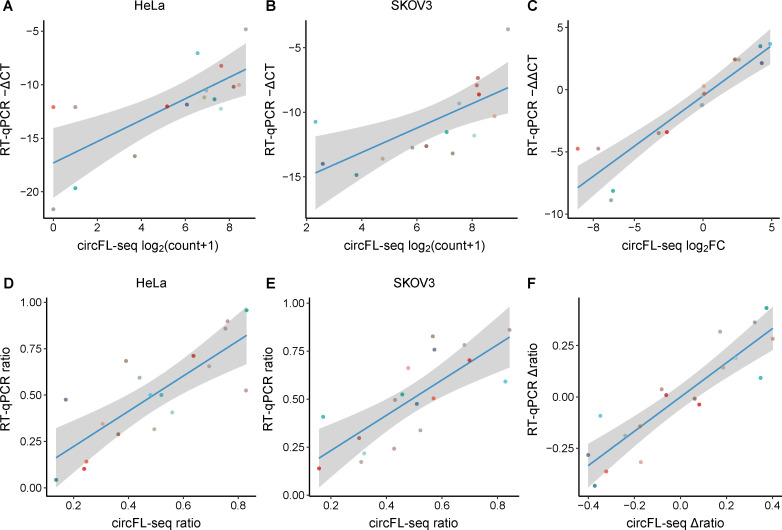
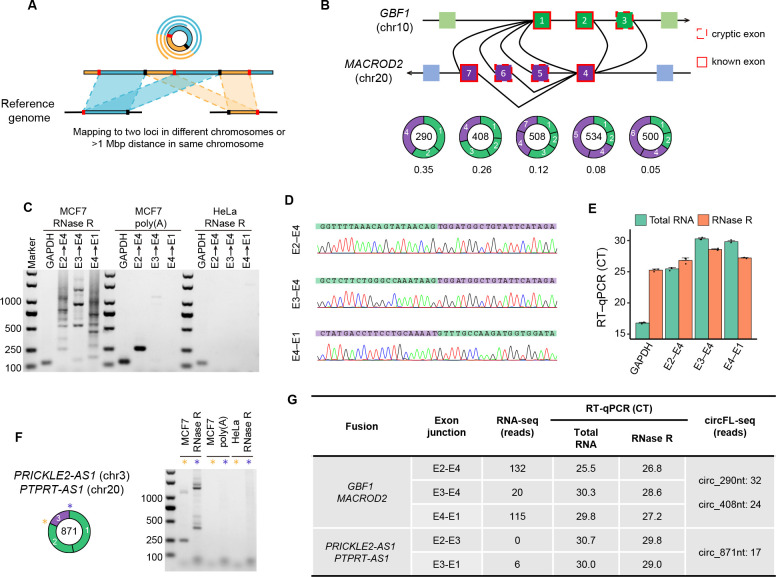
Circular RNAs (circRNAs) act through multiple mechanisms via their sequence features to fine-tune gene expression networks. Due to overlapping sequences with linear cognates, identifying internal sequences of circRNAs remains a challenge, which hinders a comprehensive understanding of circRNA functions and mechanisms. Here, based on rolling circular reverse transcription and nanopore sequencing, we developed circFL-seq, a full-length circRNA sequencing method, to profile circRNA at the isoform level. With a customized computational pipeline to directly identify full-length sequences from rolling circular reads, we reconstructed 77,606 high-quality circRNAs from seven human cell lines and two human tissues. circFL-seq benefits from rolling circles and long-read sequencing, and the results showed more than tenfold enrichment of circRNA reads and advantages for both detection and quantification at the isoform level compared to those for short-read RNA sequencing. The concordance of the RT-qPCR and circFL-seq results for the identification of differential alternative splicing suggested wide application prospects for functional studies of internal variants in circRNAs. Moreover, the detection of fusion circRNAs at the omics scale may further expand the application of circFL-seq. Taken together, the accurate identification and quantification of full-length circRNAs make circFL-seq a potential tool for large-scale screening of functional circRNAs.
环形 RNA(circRNAs)通过其序列特征通过多种机制发挥作用,精细调节基因表达网络。由于与线性同源物存在重叠序列,因此识别 circRNAs 的内部序列仍然是一个挑战,这阻碍了对 circRNA 功能和机制的全面理解。在这里,我们基于滚环反转录和纳米孔测序,开发了 circFL-seq,一种全长 circRNA 测序方法,以在异构体水平上对 circRNA 进行分析。通过一个定制的计算管道,从滚环读取中直接识别全长序列,我们从七个人类细胞系和两个人类组织中重建了 77606 个高质量的 circRNAs。circFL-seq 受益于滚环和长读测序,与短读 RNA 测序相比,结果显示 circRNA 读数的富集度提高了十倍以上,并且在异构体水平上的检测和定量方面具有优势。RT-qPCR 和 circFL-seq 结果在鉴定差异剪接方面的一致性表明,circFL-seq 在 circRNA 内部变体的功能研究中有广泛的应用前景。此外,在组学水平上检测融合 circRNAs 可能会进一步扩展 circFL-seq 的应用。总之,全长 circRNAs 的准确识别和定量使 circFL-seq 成为大规模筛选功能性 circRNAs 的潜在工具。