Department of Paediatrics and Adolescent Medicine, Haukeland University Hospital, Bergen, 5021, Norway.
Department of Clinical Medicine (K1), University of Bergen, Norway.
Ann Clin Transl Neurol. 2021 Nov;8(11):2155-2165. doi: 10.1002/acn3.51470. Epub 2021 Oct 18.
To delineate the full phenotypic spectrum of BCS1L-related disease, provide better understanding of the genotype-phenotype correlations and identify reliable prognostic disease markers.
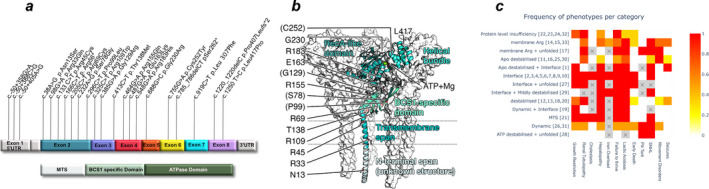
We performed a retrospective multinational cohort study of previously unpublished patients followed in 15 centres from 10 countries. Patients with confirmed biallelic pathogenic BCS1L variants were considered eligible. Clinical, laboratory, neuroimaging and genetic data were analysed. Patients were stratified into different groups based on the age of disease onset, whether homozygous or compound heterozygous for the c.232A>G (p.Ser78Gly) variant, and those with other pathogenic BCS1L variants.
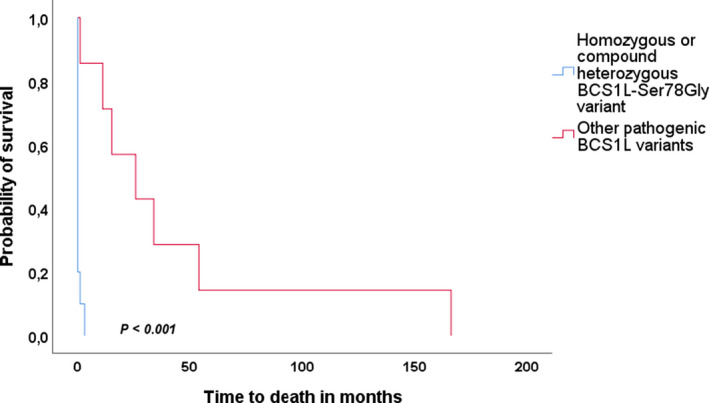
Thirty-three patients were included. We found that growth failure, lactic acidosis, tubulopathy, hepatopathy and early death were more frequent in those with disease onset within the first month of life. In those with onset after 1 month, neurological features including movement disorders and seizures were more frequent. Novel phenotypes, particularly involving movement disorder, were identified in this group. The presence of the c.232A>G (p.Ser78Gly) variant was associated with significantly worse survival and exclusively found in those with disease onset within the first month of life, whilst other pathogenic BCS1L variants were more frequent in those with later symptom onset.
The phenotypic spectrum of BCS1L-related disease comprises a continuum of clinical features rather than a set of separate syndromic clinical identities. Age of onset defines BCS1L-related disease clinically and early presentation is associated with poor prognosis. Genotype correlates with phenotype in the presence of the c.232A>G (p.Ser78Gly) variant.
描绘 BCS1L 相关疾病的全表型谱,更好地理解基因型-表型相关性,并确定可靠的预后疾病标志物。
我们对来自 10 个国家的 15 个中心的 15 例未发表的患者进行了回顾性多国队列研究。符合条件的患者为存在经证实的双等位致病性 BCS1L 变异。分析了临床、实验室、神经影像学和遗传数据。根据发病年龄、是否为 c.232A>G(p.Ser78Gly)变异的纯合子或复合杂合子,以及是否存在其他致病性 BCS1L 变异,将患者分为不同的组。
共纳入 33 例患者。我们发现,疾病在生命的第一个月内发病者更容易出现生长不良、乳酸性酸中毒、肾小管病、肝病和早期死亡;而发病在 1 个月后的患者更容易出现神经功能特征,包括运动障碍和癫痫发作。在该组中发现了新的表型,特别是涉及运动障碍的表型。c.232A>G(p.Ser78Gly)变异的存在与显著更差的生存相关,并且仅见于生命的第一个月内发病的患者,而其他致病性 BCS1L 变异在发病较晚的患者中更为常见。
BCS1L 相关疾病的表型谱包含一系列连续的临床特征,而不是一组独立的综合征性临床特征。发病年龄决定了 BCS1L 相关疾病的临床特征,早期表现与预后不良相关。在存在 c.232A>G(p.Ser78Gly)变异的情况下,基因型与表型相关。