Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington, USA.
Mass Spectrom Rev. 2023 Mar;42(2):796-821. doi: 10.1002/mas.21741. Epub 2021 Oct 31.
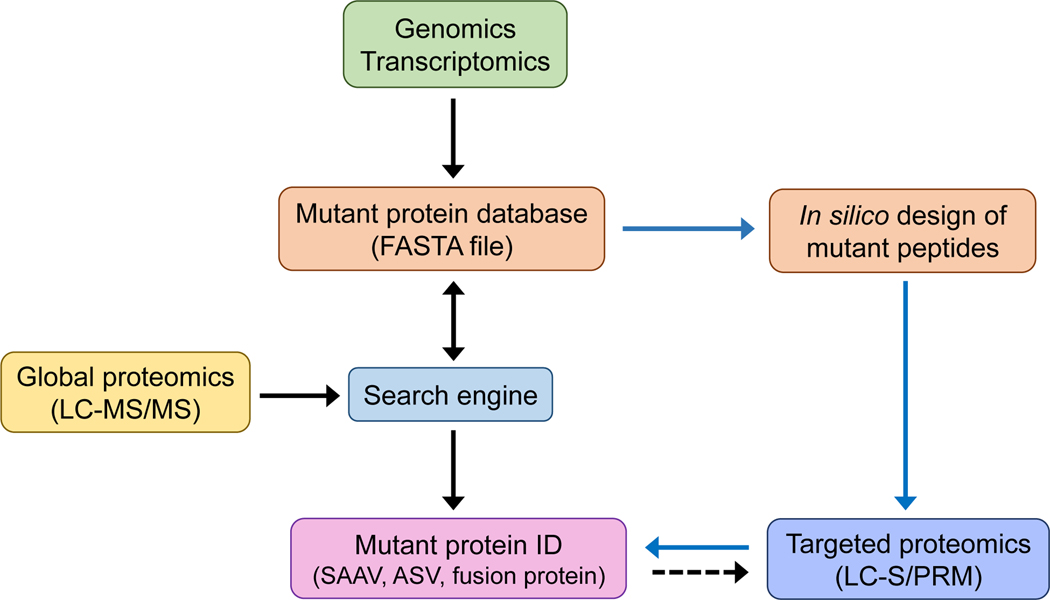
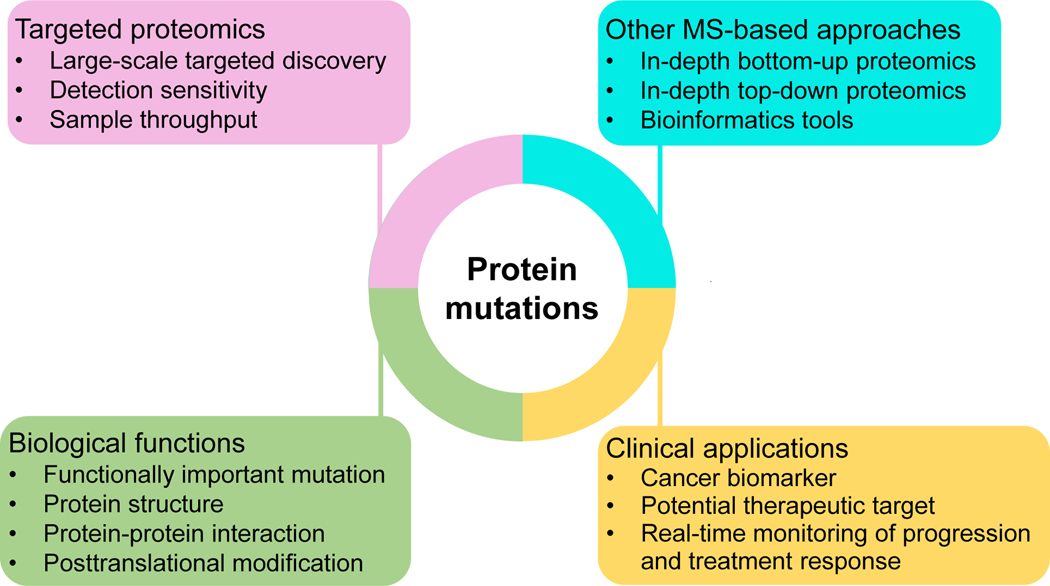
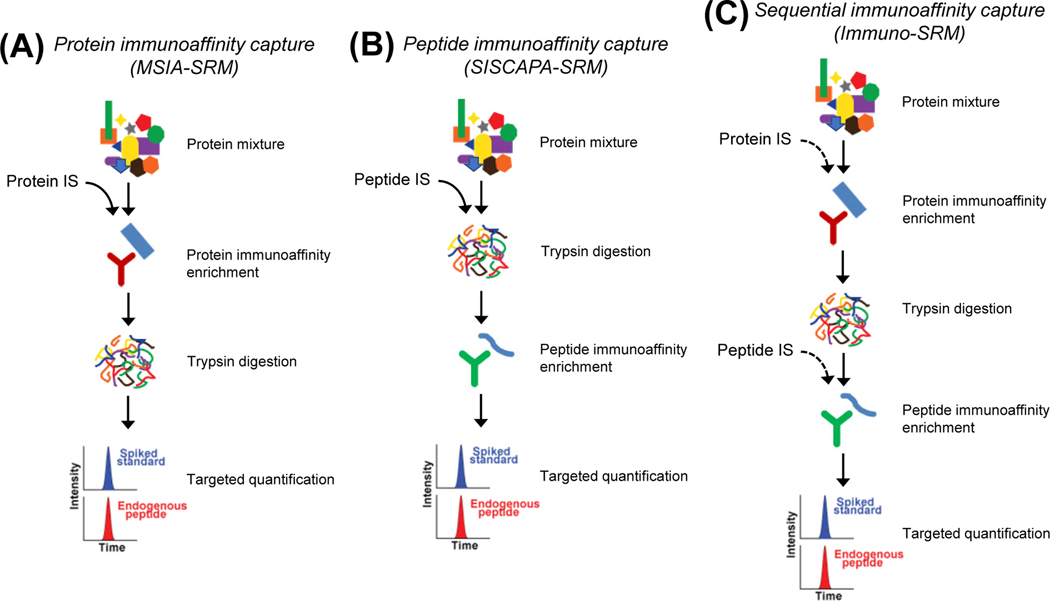
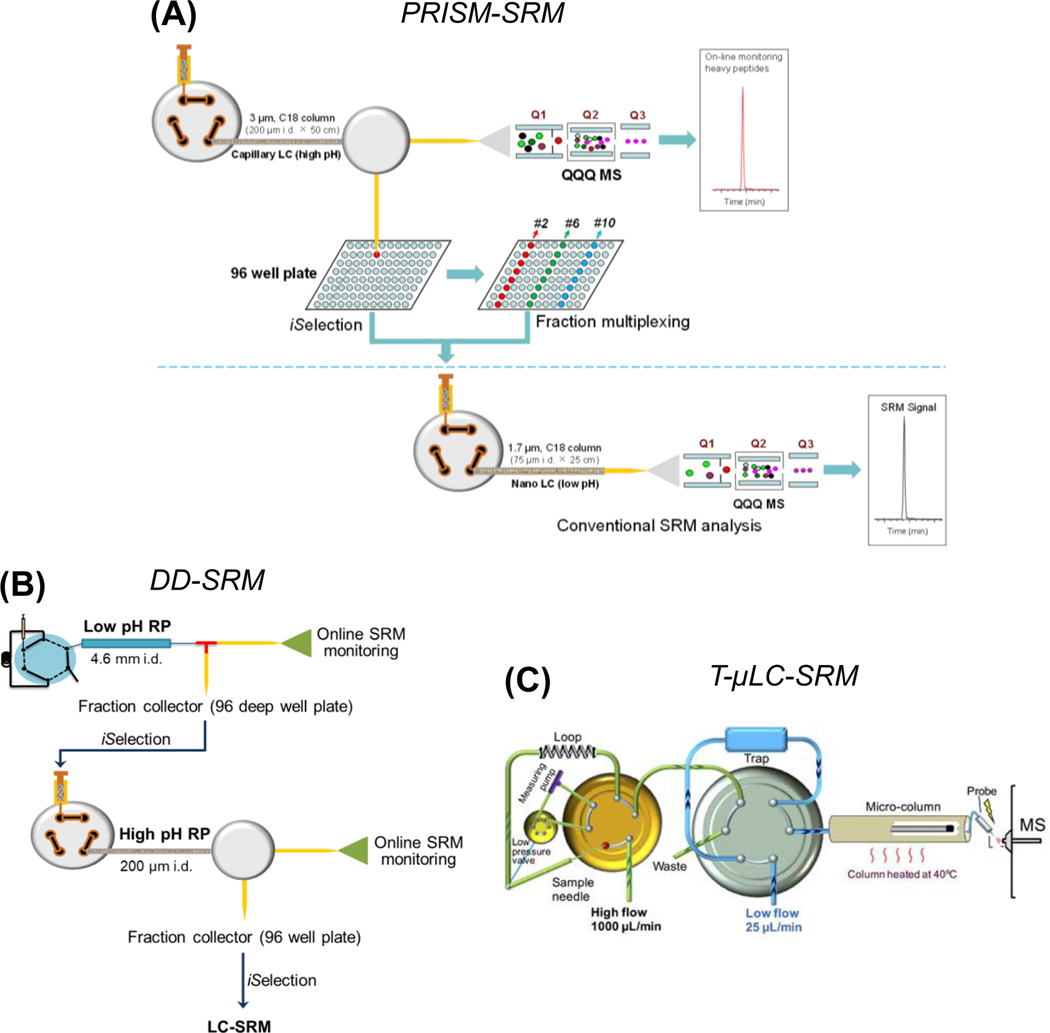
Cancers are caused by accumulated DNA mutations. This recognition of the central role of mutations in cancer and recent advances in next-generation sequencing, has initiated the massive screening of clinical samples and the identification of 1000s of cancer-associated gene mutations. However, proteomic analysis of the expressed mutation products lags far behind genomic (transcriptomic) analysis. With comprehensive global proteomics analysis, only a small percentage of single nucleotide variants detected by DNA and RNA sequencing have been observed as single amino acid variants due to current technical limitations. Proteomic analysis of mutations is important with the potential to advance cancer biomarker development and the discovery of new therapeutic targets for more effective disease treatment. Targeted proteomics using selected reaction monitoring (also known as multiple reaction monitoring) and parallel reaction monitoring, has emerged as a powerful tool with significant advantages over global proteomics for analysis of protein mutations in terms of detection sensitivity, quantitation accuracy and overall practicality (e.g., reliable identification and the scale of quantification). Herein we review recent advances in the targeted proteomics technology for enhancing detection sensitivity and multiplexing capability and highlight its broad biomedical applications for analysis of protein mutations in human bodily fluids, tissues, and cell lines. Furthermore, we review recent applications of top-down proteomics for analysis of protein mutations. Unlike the commonly used bottom-up proteomics which requires digestion of proteins into peptides, top-down proteomics directly analyzes intact proteins for more precise characterization of mutation isoforms. Finally, general perspectives on the potential of achieving both high sensitivity and high sample throughput for large-scale targeted detection and quantification of important protein mutations are discussed.
癌症是由累积的 DNA 突变引起的。这种对突变在癌症中的核心作用的认识,以及下一代测序技术的最新进展,已经启动了对临床样本的大规模筛选,并鉴定了数千种与癌症相关的基因突变。然而,突变表达产物的蛋白质组学分析远远落后于基因组学(转录组学)分析。通过全面的全局蛋白质组学分析,由于当前的技术限制,只有一小部分通过 DNA 和 RNA 测序检测到的单核苷酸变异被观察到为单个氨基酸变异。突变的蛋白质组学分析很重要,有可能推进癌症生物标志物的开发和新治疗靶点的发现,以实现更有效的疾病治疗。使用选定反应监测(也称为多重反应监测)和平行反应监测的靶向蛋白质组学,已经成为一种强大的工具,与全局蛋白质组学相比,在分析蛋白质突变方面具有显著的优势,包括检测灵敏度、定量准确性和整体实用性(例如,可靠的鉴定和定量规模)。本文综述了靶向蛋白质组学技术在提高检测灵敏度和多重检测能力方面的最新进展,并强调了其在分析人体体液、组织和细胞系中的蛋白质突变方面的广泛生物医学应用。此外,还综述了近年来自上而下的蛋白质组学在分析蛋白质突变方面的应用。与通常需要将蛋白质消化成肽的常用自下而上的蛋白质组学不同,自上而下的蛋白质组学直接分析完整的蛋白质,以更精确地表征突变异构体。最后,讨论了在大规模靶向检测和定量重要蛋白质突变时实现高灵敏度和高通量的潜力的一般观点。