The Lundquist Institute for Biomedical Innovation, Harbor-UCLA Medical Center, Torrance, United States.
Department of Biochemistry and Medical Genetics,Max Rady College of Medicine, University of Manitoba, Manitoba, Canada.
Elife. 2021 Nov 1;10:e64695. doi: 10.7554/eLife.64695.
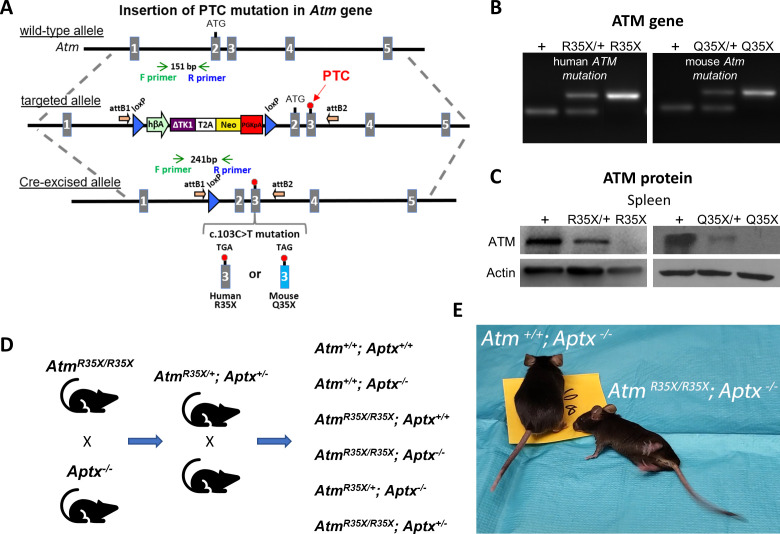
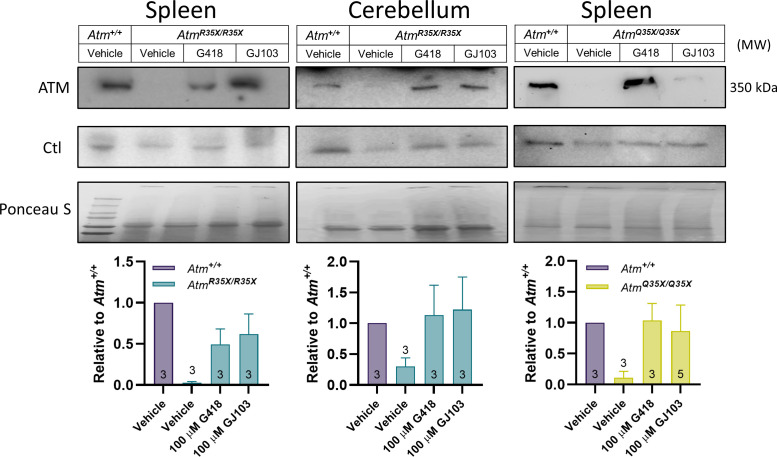
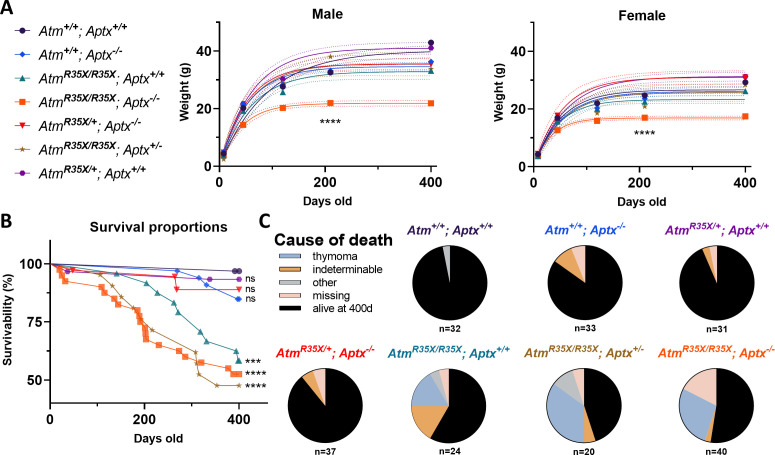
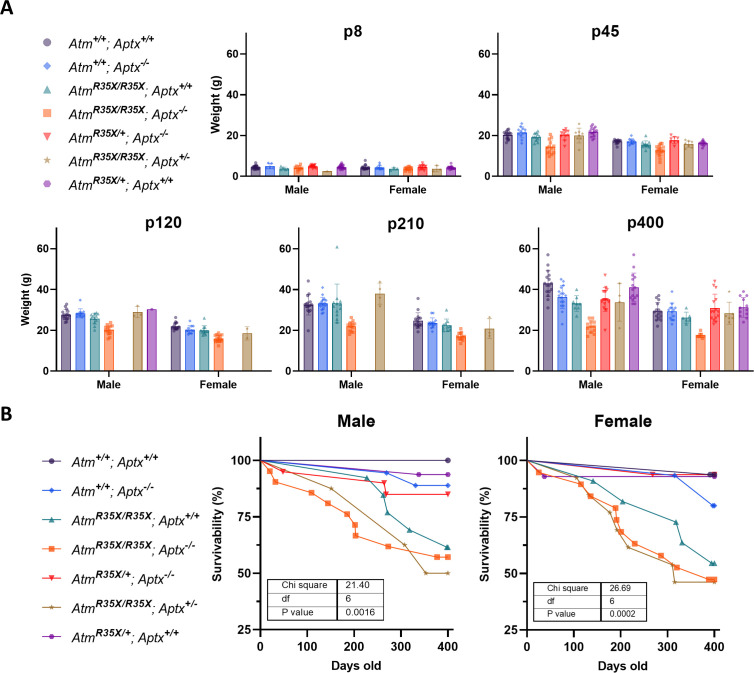
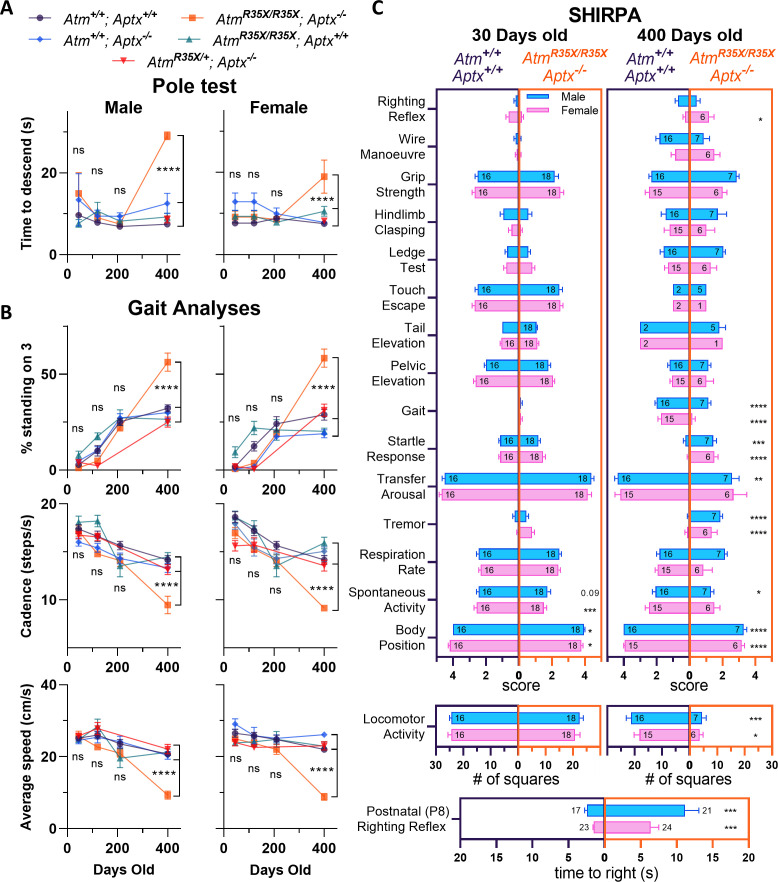
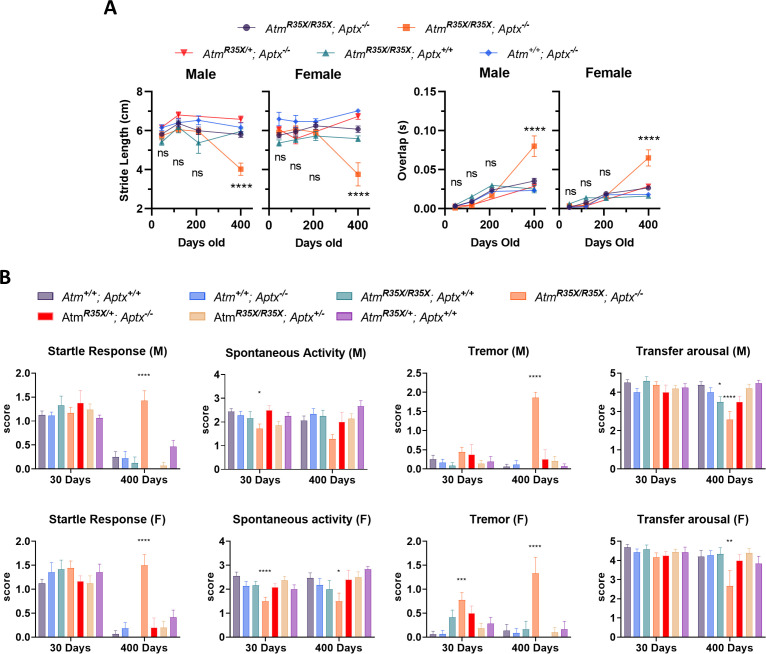
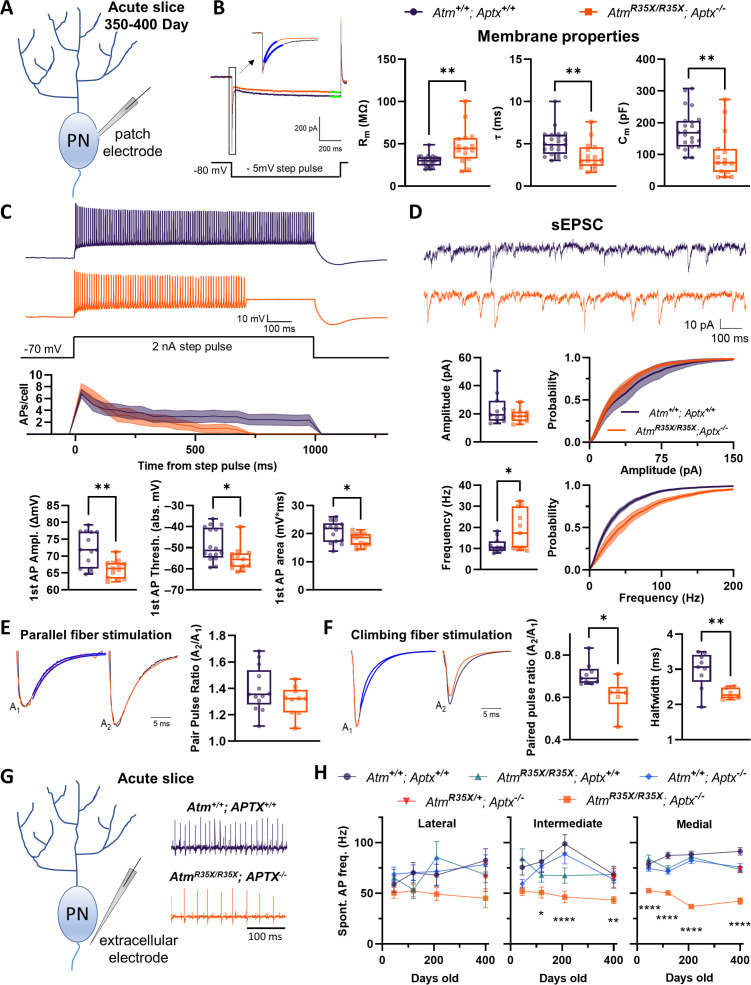
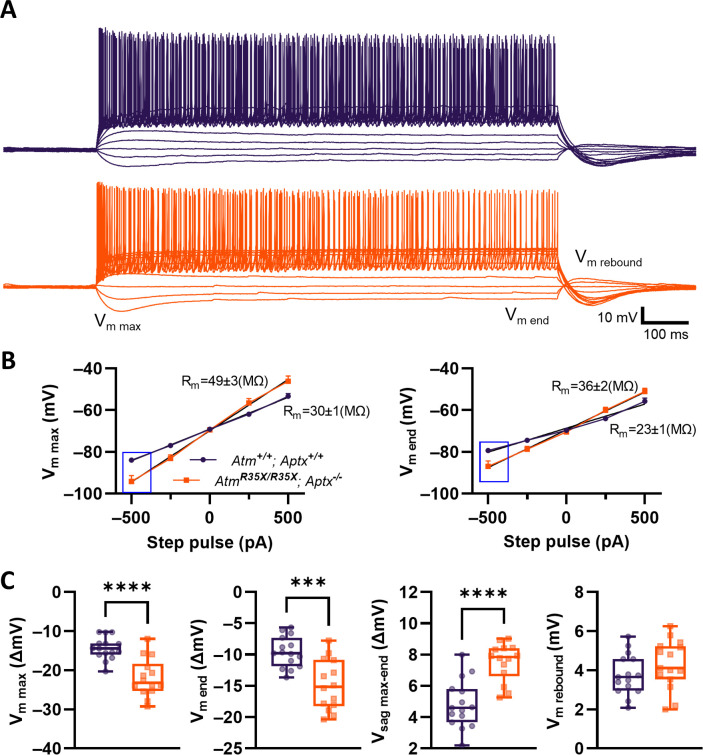
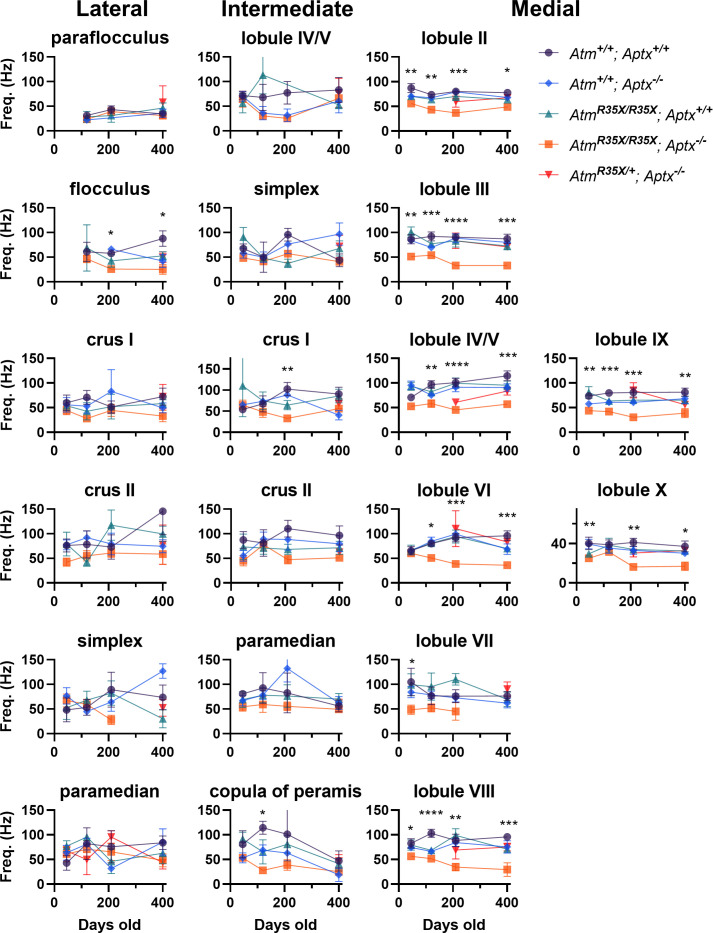
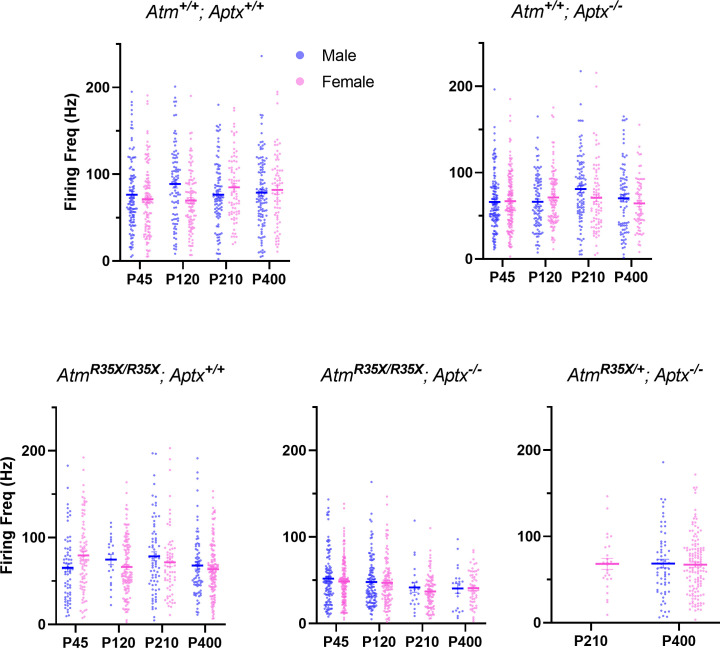
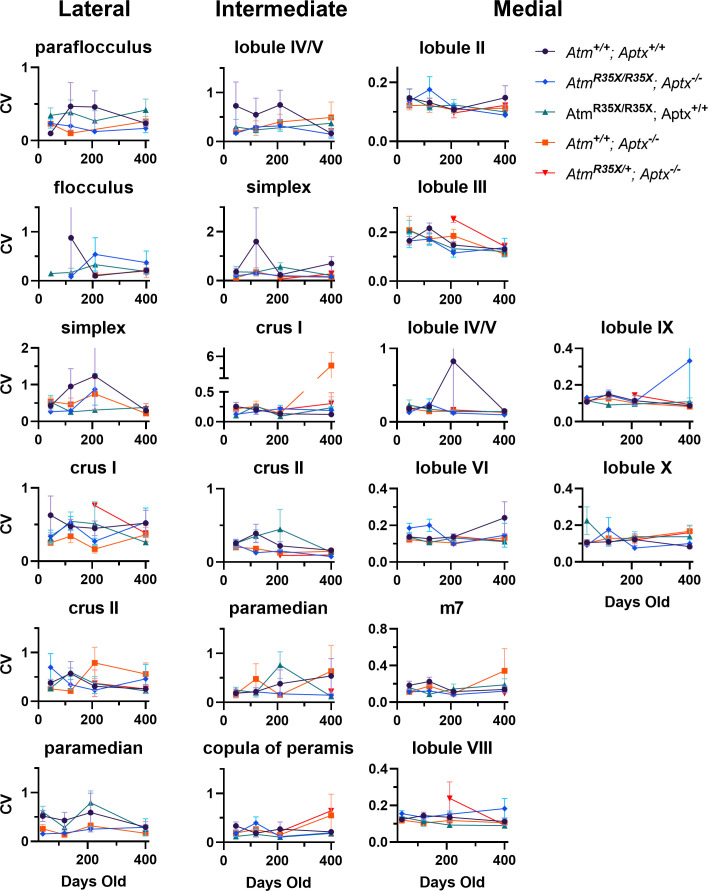
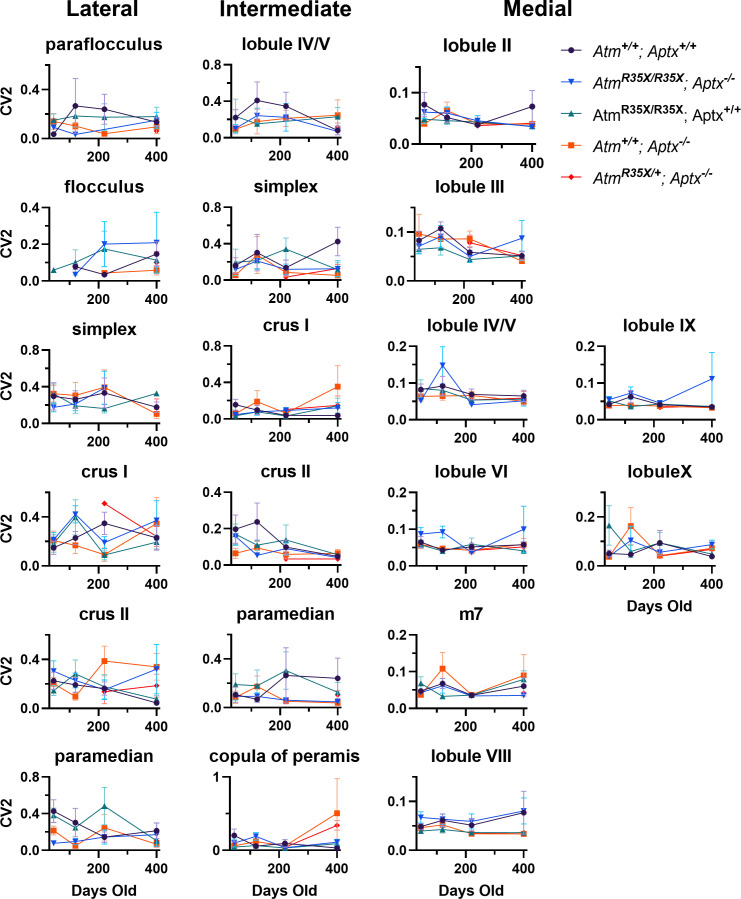
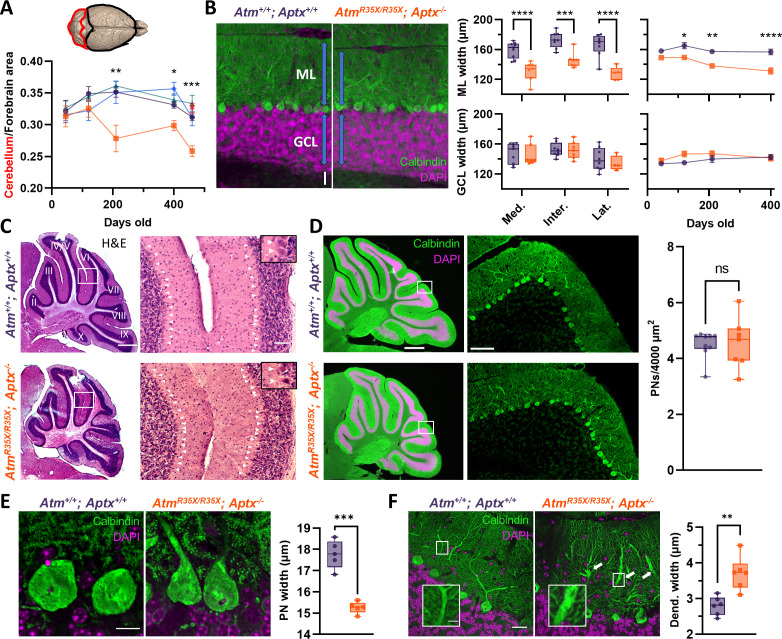
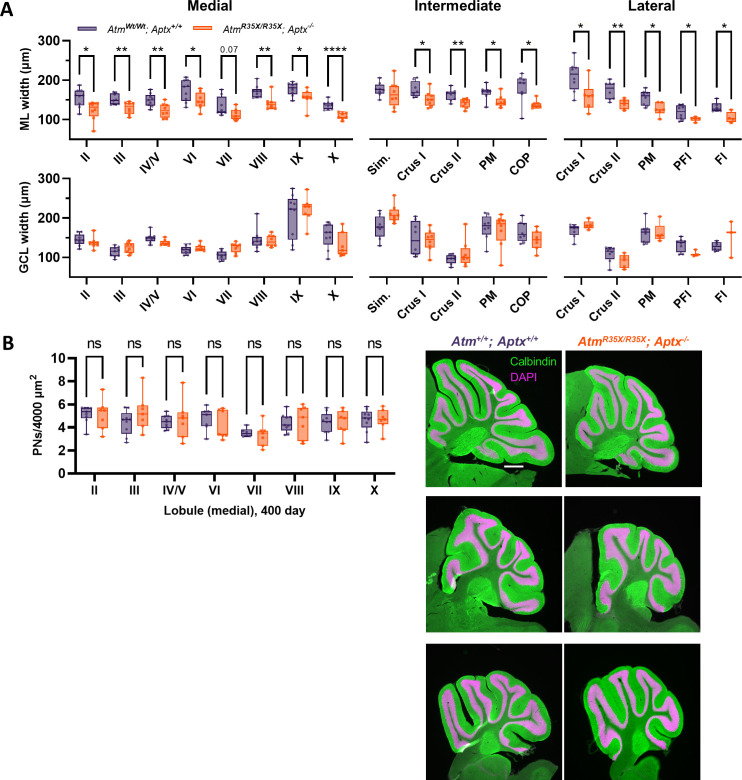
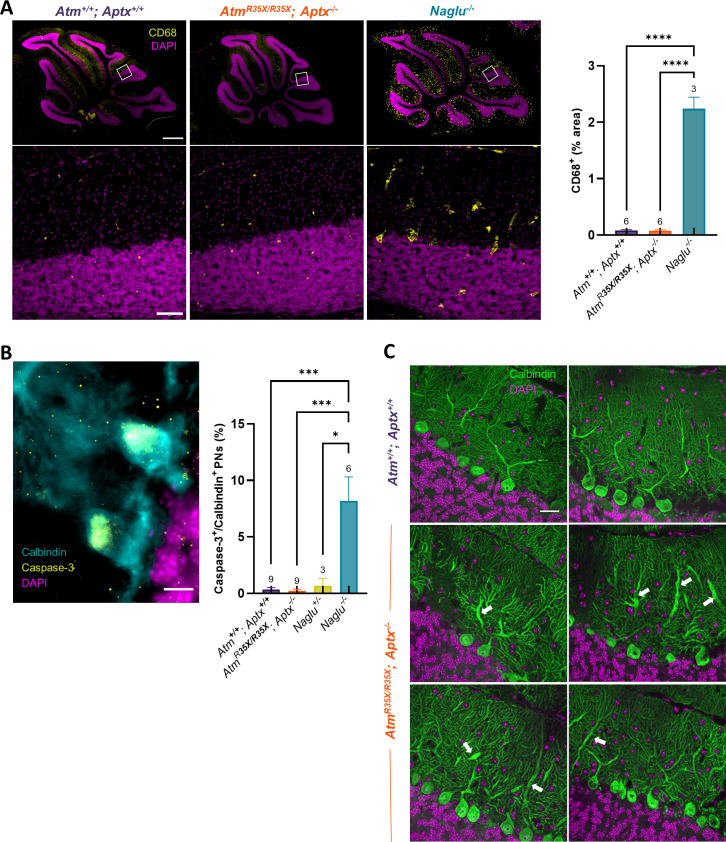
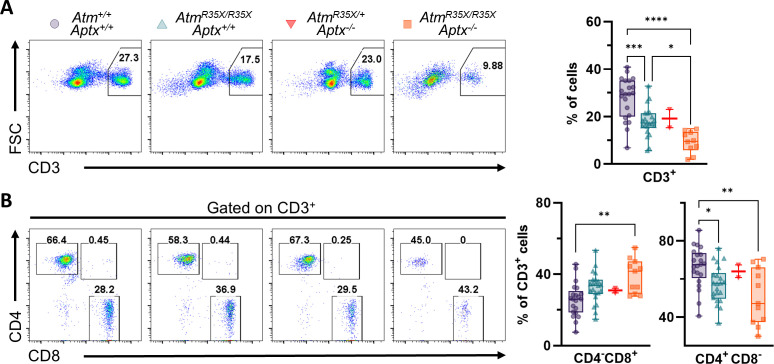
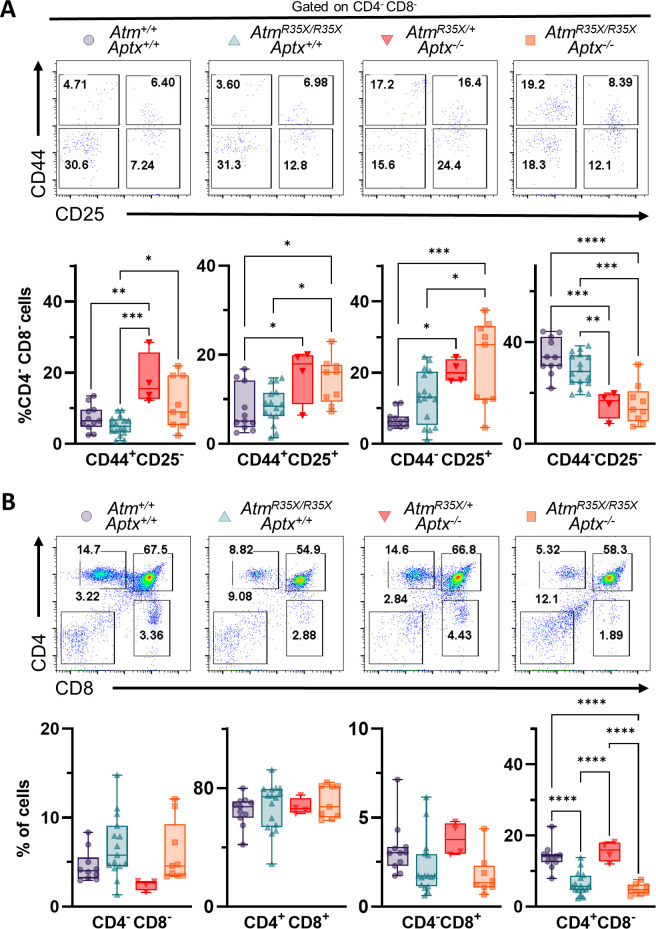
Ataxia Telangiectasia (A-T) and Ataxia with Ocular Apraxia Type 1 (AOA1) are devastating neurological disorders caused by null mutations in the genome stability genes, A-T mutated () and Aprataxin (), respectively. Our mechanistic understanding and therapeutic repertoire for treating these disorders are severely lacking, in large part due to the failure of prior animal models with similar null mutations to recapitulate the characteristic loss of motor coordination (i.e., ataxia) and associated cerebellar defects. By increasing genotoxic stress through the insertion of null mutations in both the (nonsense) and (knockout) genes in the same animal, we have generated a novel mouse model that for the first time develops a progressively severe ataxic phenotype associated with atrophy of the cerebellar molecular layer. We find biophysical properties of cerebellar Purkinje neurons (PNs) are significantly perturbed (e.g., reduced membrane capacitance, lower action potential [AP] thresholds, etc.), while properties of synaptic inputs remain largely unchanged. These perturbations significantly alter PN neural activity, including a progressive reduction in spontaneous AP firing frequency that correlates with both cerebellar atrophy and ataxia over the animal's first year of life. Double mutant mice also exhibit a high predisposition to developing cancer (thymomas) and immune abnormalities (impaired early thymocyte development and T-cell maturation), symptoms characteristic of A-T. Finally, by inserting a clinically relevant nonsense-type null mutation in , we demonstrate that mall olecule ead-hrough (SMRT) compounds can restore ATM production, indicating their potential as a future A-T therapeutic.
共济失调毛细血管扩张症(A-T)和眼运动不能伴共济失调 1 型(AOA1)是由基因组稳定性基因 AT 突变()和 Aprataxin()的无效突变引起的毁灭性神经退行性疾病。我们对这些疾病的发病机制的理解和治疗方案严重缺乏,这在很大程度上是由于之前具有相似无效突变的动物模型未能重现运动协调丧失(即共济失调)和相关小脑缺陷的特征。通过在同一动物中插入 (无义)和 (敲除)基因的无效突变来增加遗传毒性应激,我们生成了一种新型小鼠模型,该模型首次发展出与小脑分子层萎缩相关的进行性严重共济失调表型。我们发现小脑浦肯野神经元(PN)的生物物理特性受到显著干扰(例如,膜电容降低,动作电位[AP]阈值降低等),而突触输入的特性基本保持不变。这些干扰显著改变了 PN 的神经活动,包括自发性 AP 放电频率的进行性降低,这与动物一生中小脑萎缩和共济失调的发展相关。双突变小鼠还表现出易患癌症(胸腺瘤)和免疫异常(早期胸腺细胞发育和 T 细胞成熟受损)的高倾向,这些症状是 A-T 的特征。最后,通过插入临床相关的无义型无效突变,我们证明了小分子干扰(SMRT)化合物可以恢复 ATM 的产生,这表明它们可能成为未来 A-T 的治疗方法。