Department of Neurology, Erasmus University Medical Center, Dr. Molewaterplein 40, 3015 GD, Rotterdam, The Netherlands.
Department of Urology, Erasmus MC Cancer Institute, University Medical Center, Rotterdam, The Netherlands.
BMC Bioinformatics. 2021 Nov 1;22(1):535. doi: 10.1186/s12859-021-04455-3.
The FASTA file format, used to store polymeric sequence data, has become a bioinformatics file standard used for decades. The relatively large files require additional files, beyond the scope of the original format, to identify sequences and to provide random access. Multiple compressors have been developed to archive FASTA files back and forth, but these lack direct access to targeted content or metadata of the archive. Moreover, these solutions are not directly backwards compatible to FASTA files, resulting in limited software integration.
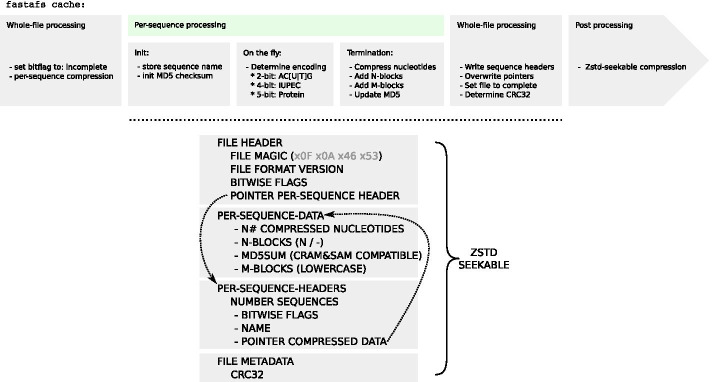
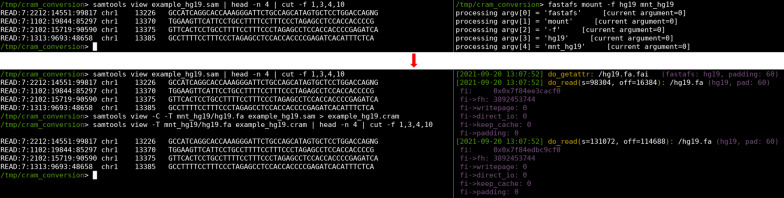
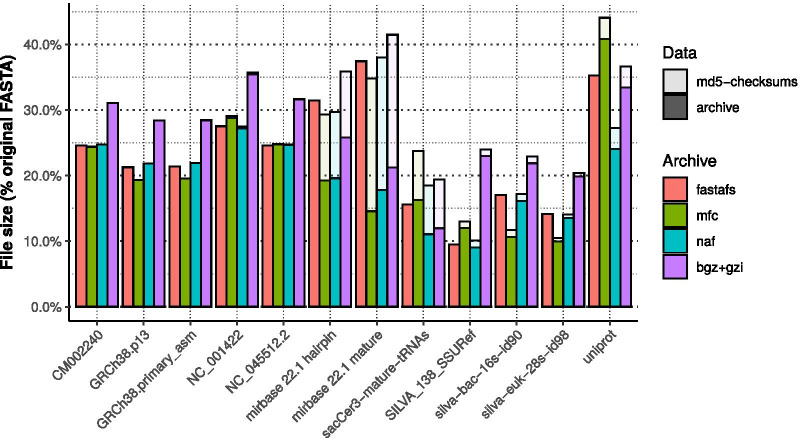
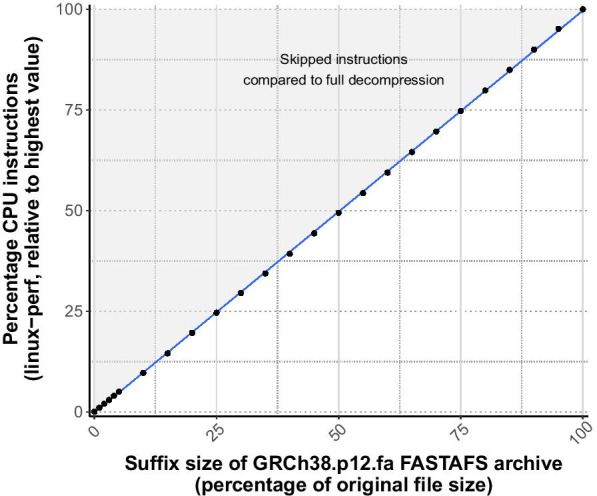
We designed a linux based toolkit that virtualises the content of DNA, RNA and protein FASTA archives into the filesystem by using filesystem in userspace. This guarantees in-sync virtualised metadata files and offers fast random-access decompression using bit encodings plus Zstandard (zstd). The toolkit, FASTAFS, can track all its system-wide running instances, allows file integrity verification and can provide, instantly, scriptable access to sequence files and is easy to use and deploy. The file compression ratios were comparable but not superior to other state of the art archival tools, despite the innovative random access feature implemented in FASTAFS.
FASTAFS is a user-friendly and easy to deploy backwards compatible generic purpose solution to store and access compressed FASTA files, since it offers file system access to FASTA files as well as in-sync metadata files through file virtualisation. Using virtual filesystems as in-between layer offers format conversion without the need to rewrite code into different programming languages while preserving compatibility.
FASTA 文件格式,用于存储聚合序列数据,已成为几十年来生物信息学文件的标准。相对较大的文件需要额外的文件来识别序列并提供随机访问,超出了原始格式的范围。已经开发了多种压缩器来来回回地归档 FASTA 文件,但这些压缩器缺乏对存档内容或元数据的直接访问。此外,这些解决方案与 FASTA 文件不直接向后兼容,导致软件集成有限。
我们设计了一个基于 Linux 的工具包,通过在用户空间中使用文件系统,将 DNA、RNA 和蛋白质 FASTA 档案的内容虚拟化为文件系统。这保证了同步的虚拟化元数据文件,并通过位编码加 Zstandard(zstd)提供快速随机访问解压缩。FASTAFS 工具包可以跟踪其所有系统范围内的运行实例,允许文件完整性验证,并可以即时提供对序列文件的脚本访问,并且易于使用和部署。尽管 FASTAFS 实现了创新的随机访问功能,但文件压缩比与其他最先进的归档工具相当,但不占优势。
FASTAFS 是一种用户友好且易于部署的向后兼容的通用解决方案,用于存储和访问压缩的 FASTA 文件,因为它通过文件虚拟化为 FASTA 文件以及同步的元数据文件提供文件系统访问。使用虚拟文件系统作为中间层,在不将代码重写为不同编程语言的情况下提供格式转换,同时保持兼容性。