Peters Kathryn W, Gong Xiaoyan, Frizzell Raymond A
Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States.
Front Physiol. 2021 Oct 26;12:695767. doi: 10.3389/fphys.2021.695767. eCollection 2021.
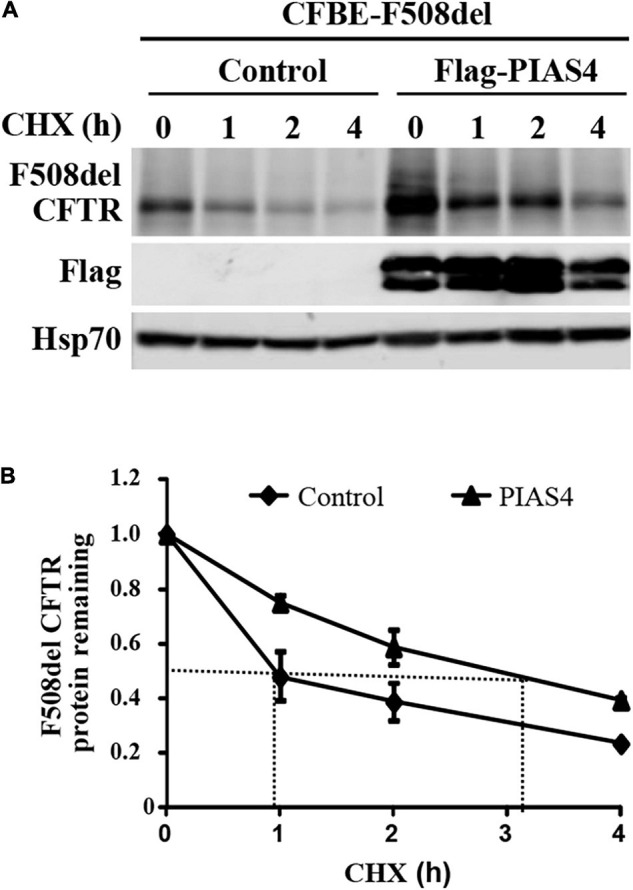
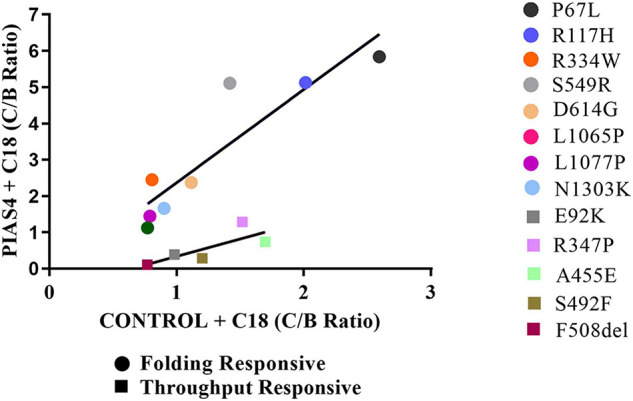
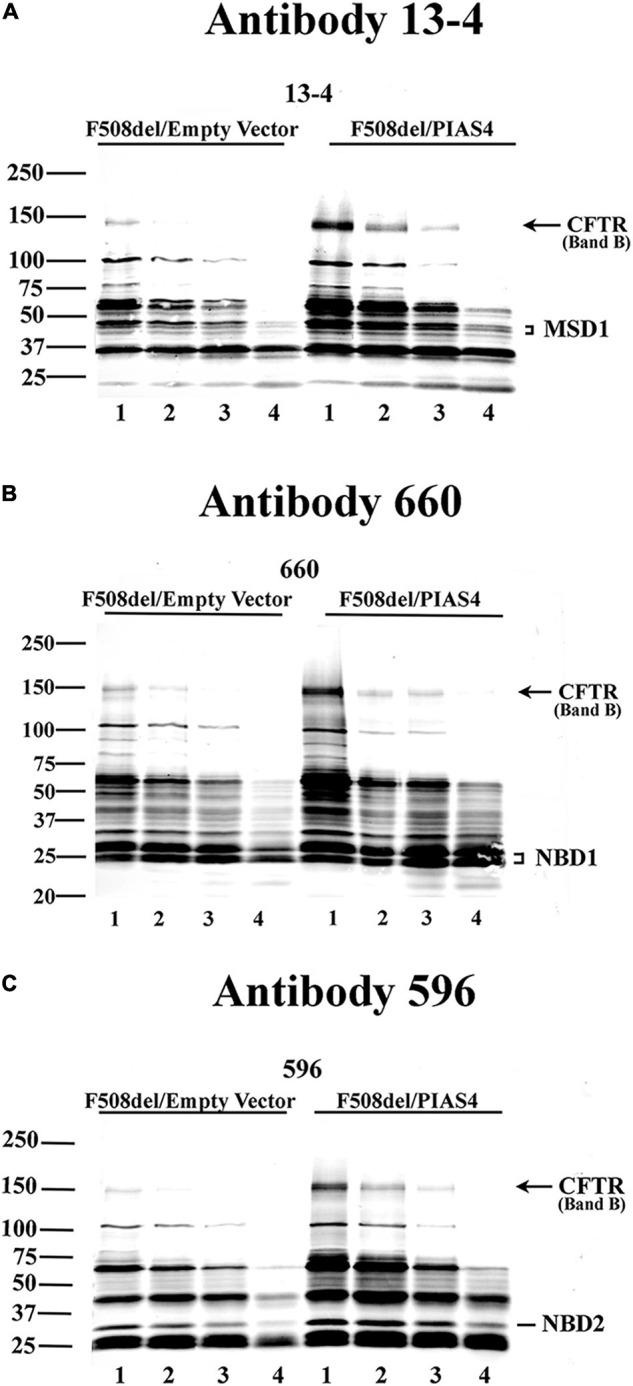
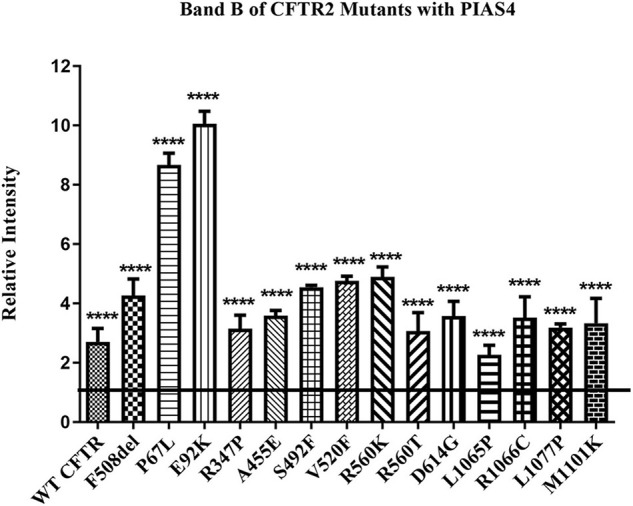
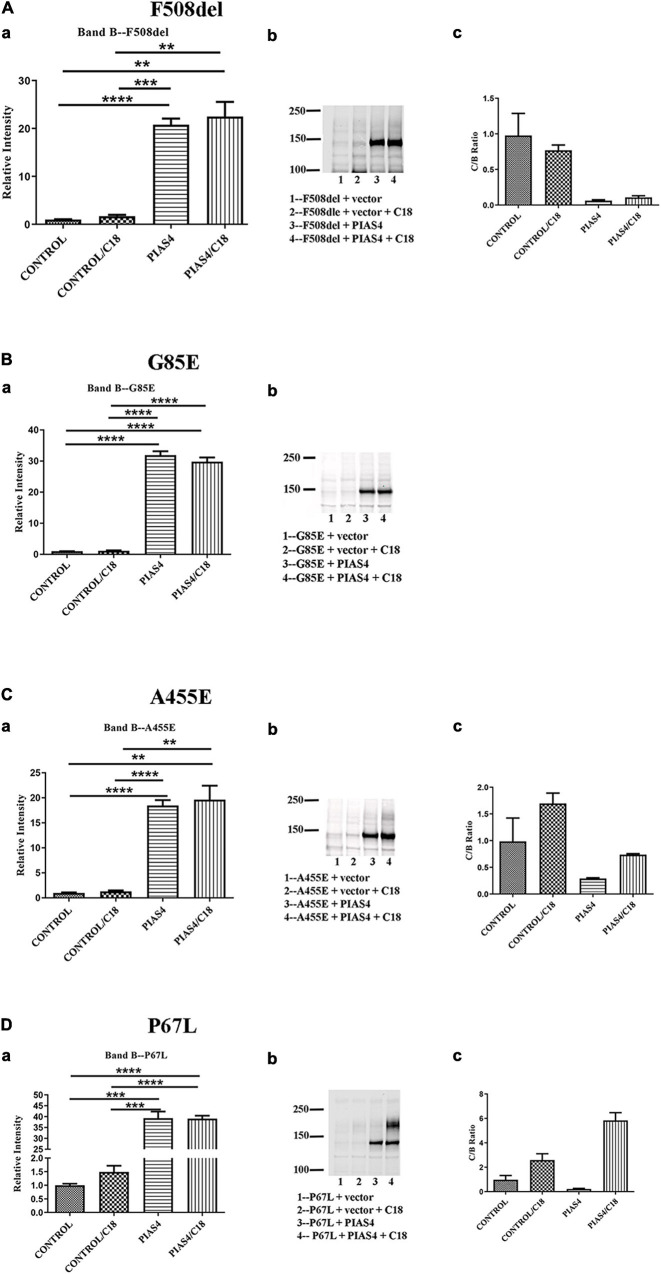
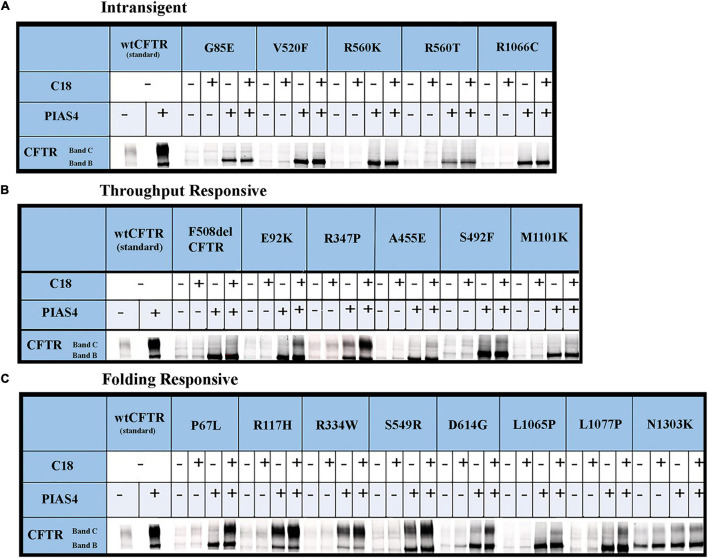
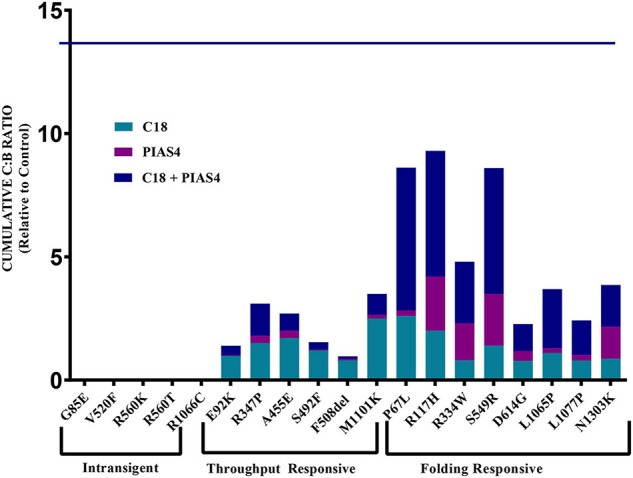
Most cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that lead to protein misfolding and degradation by the ubiquitin-proteasome system. Previous studies demonstrated that PIAS4 facilitates the modification of wild-type (WT) and F508del CFTR by small ubiquitin-like modifier (SUMO)-1, enhancing CFTR biogenesis by slowing immature CFTR degradation and producing increased immature CFTR band B. We evaluated two correction strategies using misfolding mutants, including the common variant, F508del. We examined the effects on mutant expression of co-expression with PIAS4 (E3 SUMO ligase), and/or the corrector, C18. To study the impact of these correction conditions, we transfected CFBE410- cells, a bronchial epithelial cell line, with a CFTR mutant plus: (1) empty vector, (2) empty vector plus overnight 5 μM C18, (3) PIAS4, and (4) PIAS4 plus C18. We assessed expression at steady state by immunoblot of CFTR band B, and if present, band C, and the corresponding C:B band ratio. The large PIAS4-induced increase in band B expression allowed us to ask whether C18 could act on the now abundant immature protein to enhance correction above the control level, as reported by the C:B ratio. The data fell into three mutant CFTR categories as follows: (1) intransigent: no observable band C under any condition (i.e., C:B = 0); (2) throughput responsive: a C:B ratio less than control, but suggesting that the increased band C resulted from PIAS4-induced increases in band B production; and (3) folding responsive: a C:B ratio greater than control, reflecting C18-induced folding greater than that expected from increased throughput due to the PIAS4-induced band B level. These results suggest that the immature forms of CFTR folding intermediates occupy different loci within the energetic/kinetic folding landscape of CFTR. The evaluation of their properties could assist in the development of correctors that can target the more difficult-to-fold mutant conformations that occupy different sites within the CFTR folding pathway.
大多数囊性纤维化是由囊性纤维化跨膜传导调节因子(CFTR)基因突变引起的,这些突变会导致蛋白质错误折叠并被泛素 - 蛋白酶体系统降解。先前的研究表明,PIAS4促进小泛素样修饰物(SUMO)-1对野生型(WT)和F508del CFTR的修饰,通过减缓未成熟CFTR的降解来增强CFTR生物合成,并产生增加的未成熟CFTR条带B。我们使用错误折叠突变体评估了两种校正策略,包括常见变体F508del。我们研究了与PIAS4(E3 SUMO连接酶)和/或校正剂C18共表达对突变体表达的影响。为了研究这些校正条件的影响,我们用CFTR突变体转染支气管上皮细胞系CFBE410 - 细胞,并分别添加:(1)空载体,(2)空载体加5μM C18过夜,(3)PIAS4,以及(4)PIAS4加C18。我们通过对CFTR条带B(若存在则包括条带C)进行免疫印迹以及相应的C:B条带比率来评估稳态下的表达。PIAS4诱导的条带B表达大幅增加使我们能够探究C18是否可以作用于现在大量存在的未成熟蛋白,以将校正增强至对照水平以上,如C:B比率所示。数据分为以下三类突变型CFTR:(1)顽固型:在任何条件下均未观察到条带C(即C:B = 0);(2)通量响应型:C:B比率低于对照,但表明增加的条带C是由PIAS4诱导的条带B产生增加所致;(3)折叠响应型:C:B比率高于对照,反映C18诱导的折叠大于因PIAS4诱导的条带B水平导致的通量增加所预期的折叠。这些结果表明,CFTR折叠中间体的未成熟形式在CFTR的能量/动力学折叠格局中占据不同位点。对其特性的评估有助于开发能够靶向CFTR折叠途径中占据不同位点的更难折叠突变体构象的校正剂。