Gale Jenna R, Kosobucki Gabrielle J, Hartnett-Scott Karen A, Aizenman Elias
Department of Neurobiology and Pittsburgh Institute for Neurodegenerative Diseases, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States.
Front Pharmacol. 2021 Oct 28;12:773455. doi: 10.3389/fphar.2021.773455. eCollection 2021.
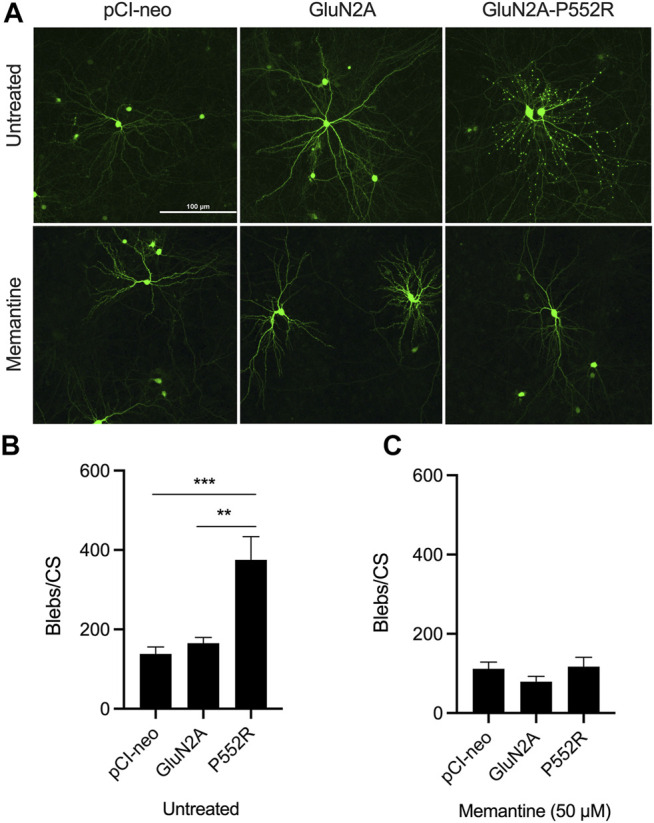
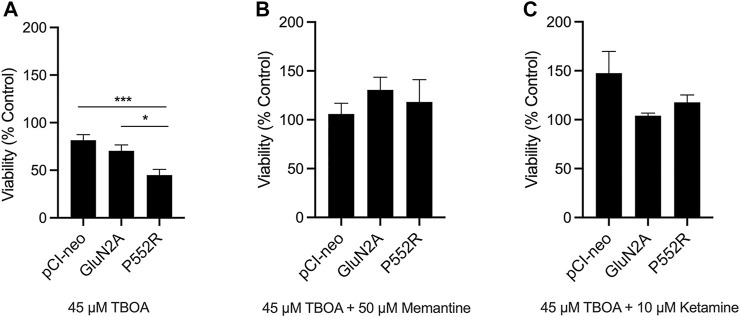
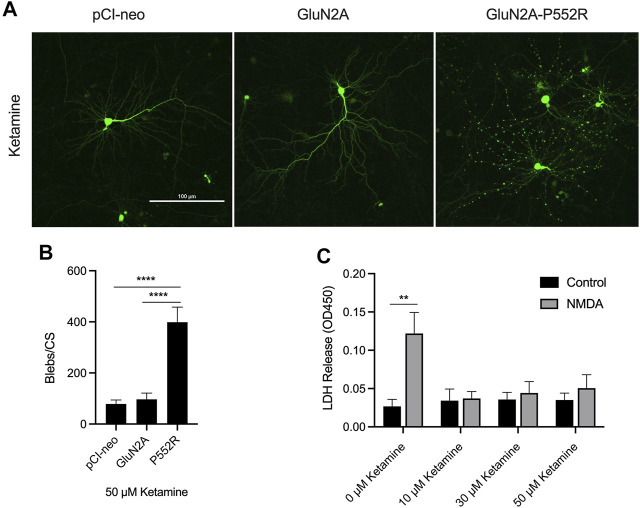
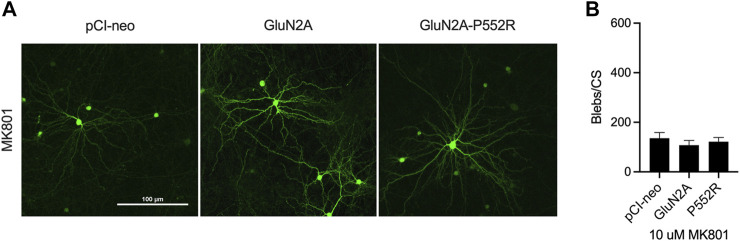
Mutations in N-methyl-d-aspartate receptors (NMDAR) subunits have been implicated in a growing number of human neurodevelopmental disorders. Previously, a mutation in , encoding the GluN2A subunit, was identified in a patient with severe epilepsy and developmental delay. This missense mutation, which leads to GluN2A-P552R, produces significant dendrotoxicity in transfected rodent cortical neurons, as evidenced by pronounced dendritic blebbing. This injurious process can be prevented by treatment with the NMDA antagonist memantine. Given the increasing use of FDA approved NMDA antagonists to treat patients with mutations, who may have seizures refractory to traditional anti-epileptic drugs, we investigated whether additional NMDA antagonists were effective in attenuating neurotoxicity associated with GluN2A-P552R expression. Intriguingly, we found that while treatment with memantine can effectively block GluN2A-P552R-mediated dendrotoxicity, treatment with ketamine does not, despite the fact that both drugs work as open NMDAR channel blockers. Interestingly, we found that neurons expressing GluN2A-P552R were more vulnerable to an excitotoxic insult-an effect that, in this case, could be equally rescued by both memantine and ketamine. These findings suggest that GluN2A-P552R induced dendrotoxicity and increased vulnerability to excitotoxic stress are mediated through two distinct mechanisms. The differences between memantine and ketamine in halting GluN2A-P552R dendrotoxicity could not be explained by NMDA antagonist induced changes in MAP or Src kinase activation, previously shown to participate in NMDA-induced excitotoxicity. Our findings strongly suggest that not all NMDA antagonists may be of equal clinical utility in treating -mediated neurological disorders, despite a shared mechanism of action.
N-甲基-D-天冬氨酸受体(NMDAR)亚基突变与越来越多的人类神经发育障碍有关。此前,在一名患有严重癫痫和发育迟缓的患者中,发现了编码GluN2A亚基的基因突变。这种错义突变导致GluN2A-P552R,在转染的啮齿动物皮质神经元中产生显著的树突毒性,明显的树突肿胀就是证据。用NMDA拮抗剂美金刚治疗可以预防这种损伤过程。鉴于FDA批准的NMDA拮抗剂越来越多地用于治疗有基因突变、可能对传统抗癫痫药物难治性癫痫发作的患者,我们研究了其他NMDA拮抗剂是否能有效减轻与GluN2A-P552R表达相关的神经毒性。有趣的是,我们发现虽然用美金刚治疗可以有效阻断GluN2A-P552R介导的树突毒性,但氯胺酮治疗却不能,尽管这两种药物都作为开放的NMDAR通道阻滞剂起作用。有趣的是,我们发现表达GluN2A-P552R的神经元对兴奋性毒性损伤更敏感——在这种情况下,美金刚和氯胺酮都能同样挽救这种效应。这些发现表明,GluN2A-P552R诱导的树突毒性和对兴奋性毒性应激易感性增加是通过两种不同机制介导的。美金刚和氯胺酮在阻止GluN2A-P552R树突毒性方面的差异,不能用NMDA拮抗剂诱导的MAP或Src激酶激活变化来解释,此前已表明这些变化参与NMDA诱导的兴奋性毒性。我们的研究结果强烈表明,尽管作用机制相同,但并非所有NMDA拮抗剂在治疗介导的神经系统疾病中都具有同等的临床效用。