Helsinki Institute for Information Technology HIIT, Department of Mathematics and Statistics, University of Helsinki, Helsinki, Finland.
Department of Biostatistics, University of Oslo, Oslo, Norway.
Microb Genom. 2021 Nov;7(11). doi: 10.1099/mgen.0.000691.
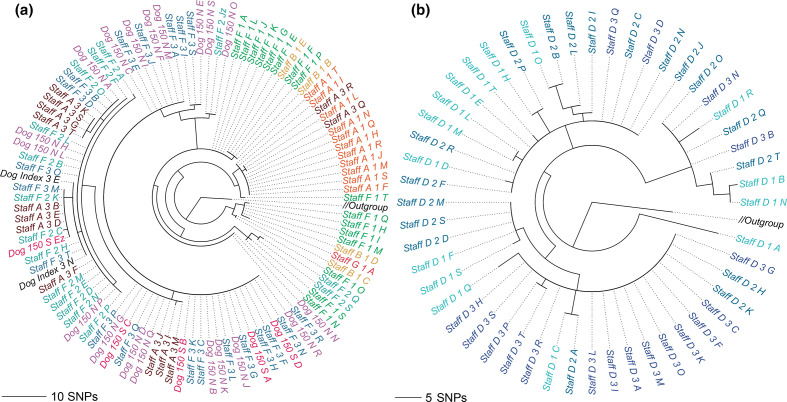
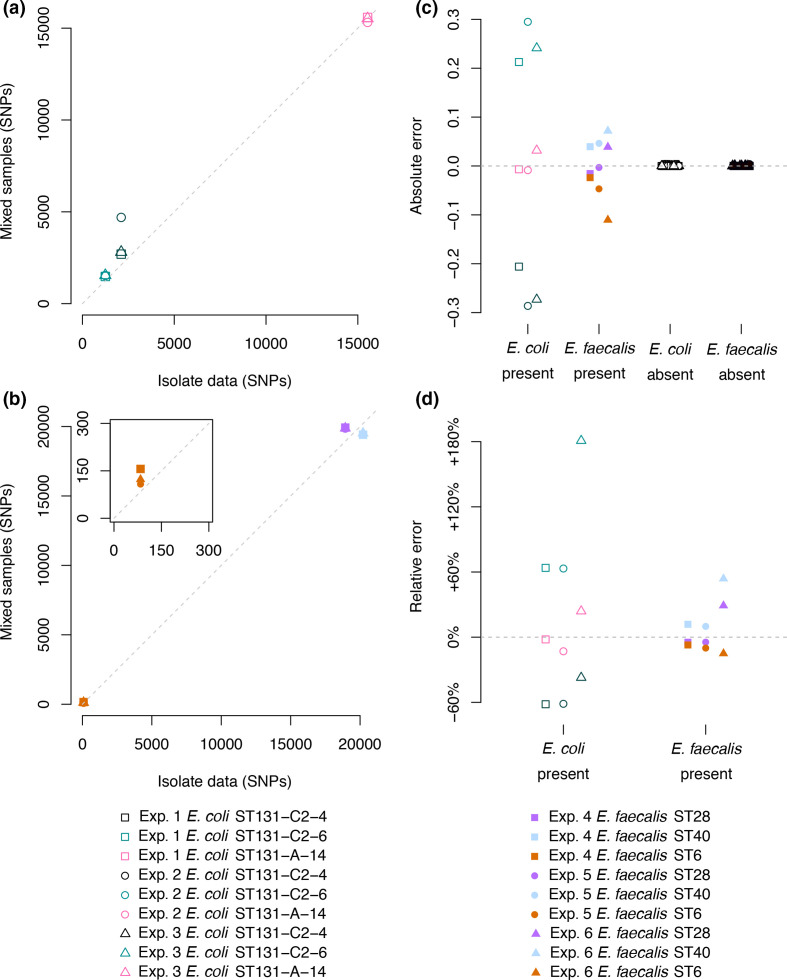
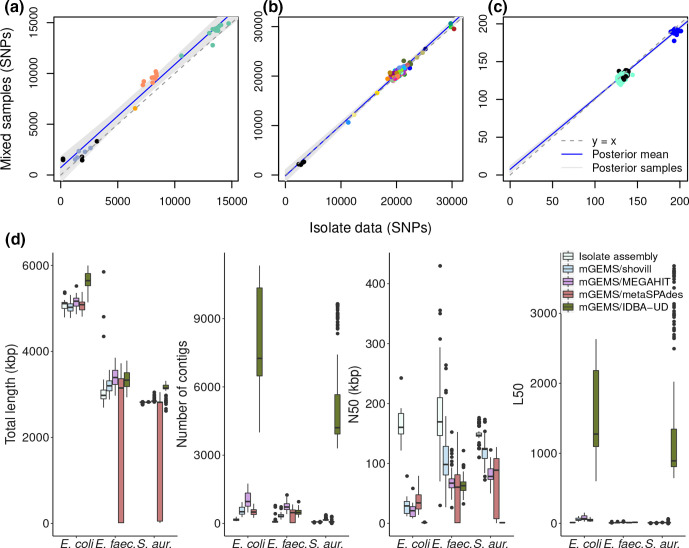
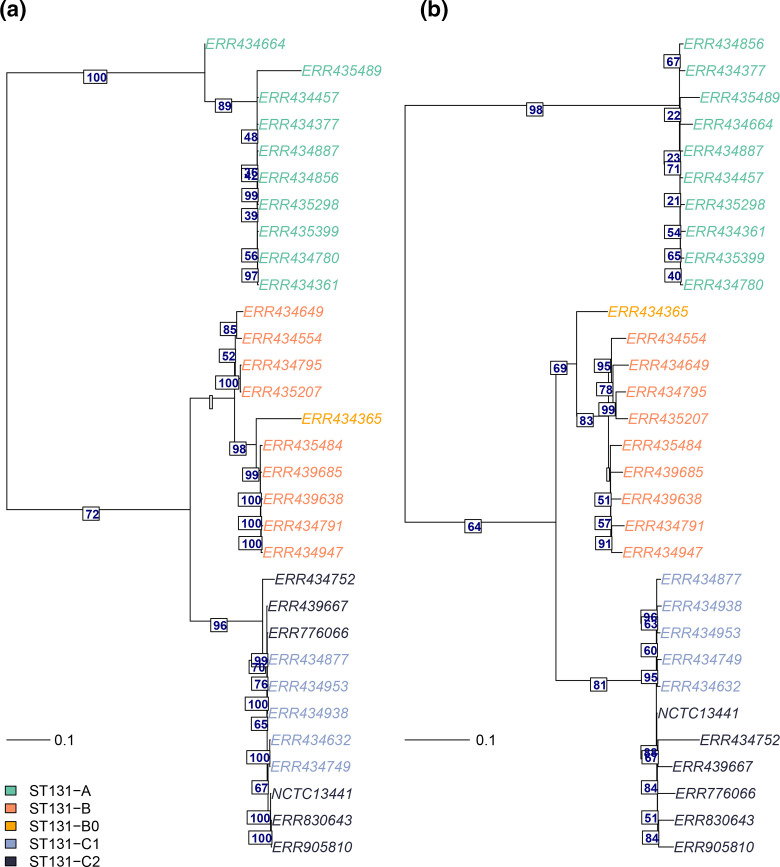
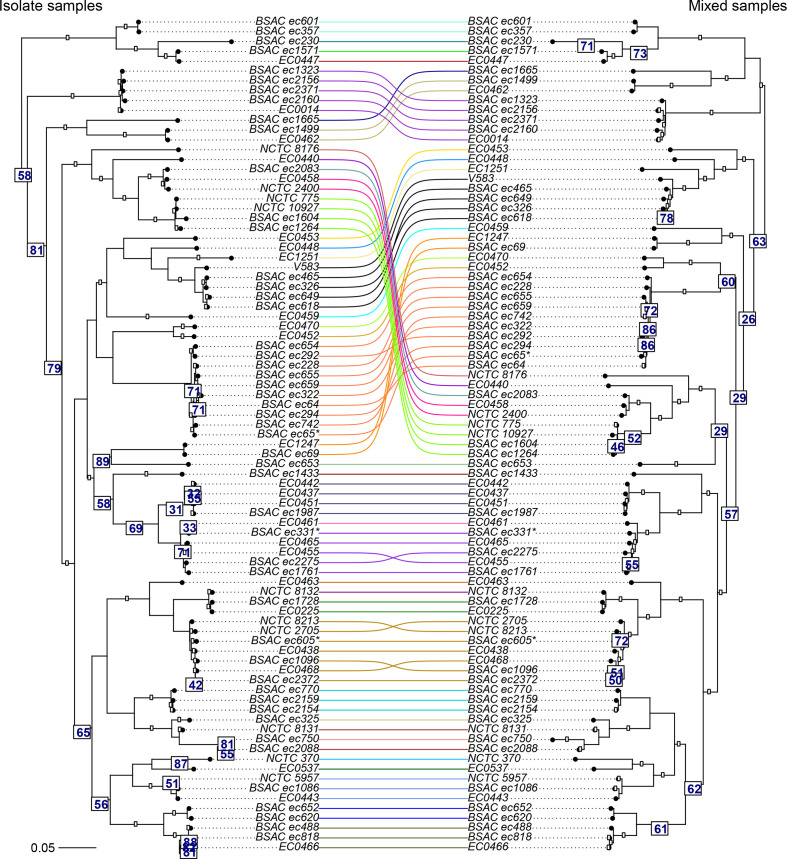
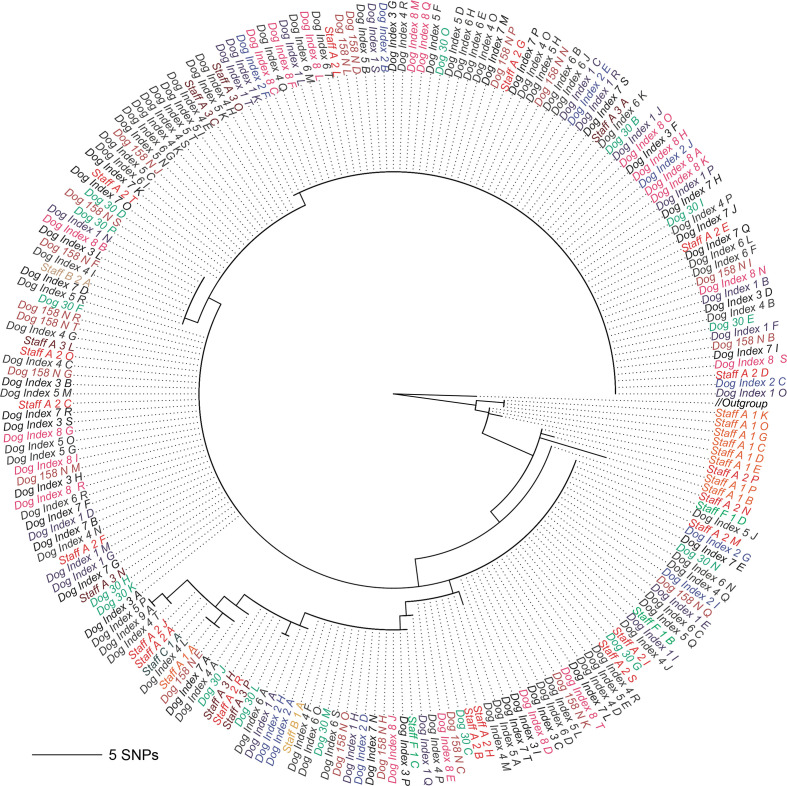
Genomic epidemiology is a tool for tracing transmission of pathogens based on whole-genome sequencing. We introduce the mGEMS pipeline for genomic epidemiology with plate sweeps representing mixed samples of a target pathogen, opening the possibility to sequence all colonies on selective plates with a single DNA extraction and sequencing step. The pipeline includes the novel mGEMS read binner for probabilistic assignments of sequencing reads, and the scalable pseudoaligner Themisto. We demonstrate the effectiveness of our approach using closely related samples in a nosocomial setting, obtaining results that are comparable to those based on single-colony picks. Our results lend firm support to more widespread consideration of genomic epidemiology with mixed infection samples.
基因组流行病学是一种基于全基因组测序追踪病原体传播的工具。我们引入了 mGEMS 基因组流行病学管道,该管道使用平板扫荡来代表目标病原体的混合样本,从而有可能通过单个 DNA 提取和测序步骤对选择性平板上的所有菌落进行测序。该管道包括新颖的 mGEMS 读取分配器,用于对测序读取进行概率分配,以及可扩展的伪对齐器 Themisto。我们使用医院环境中的密切相关的样本证明了我们方法的有效性,得到的结果与基于单菌落挑选的结果相当。我们的结果为更广泛地考虑混合感染样本的基因组流行病学提供了有力支持。