Vulcan Biosciences, Birmingham, Alabama, USA.
University of California at Davis Proteomics Core, Davis, California, USA.
Mol Cell Proteomics. 2022 Jan;21(1):100180. doi: 10.1016/j.mcpro.2021.100180. Epub 2021 Nov 20.
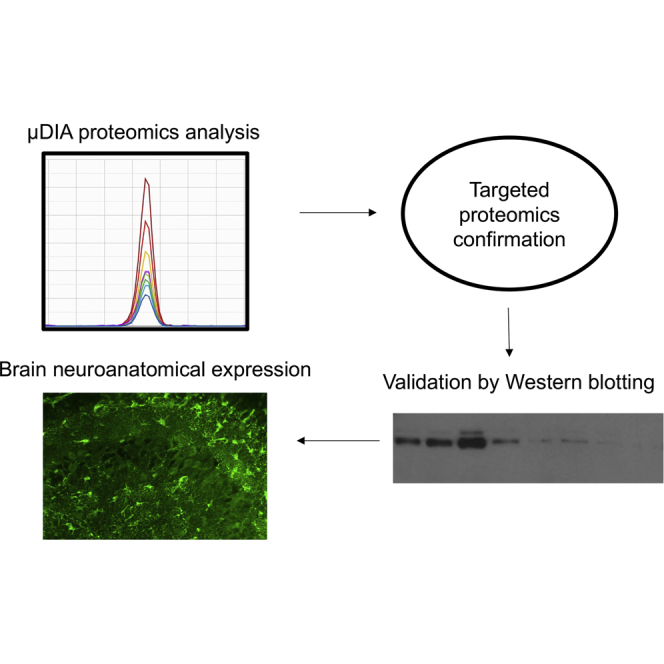
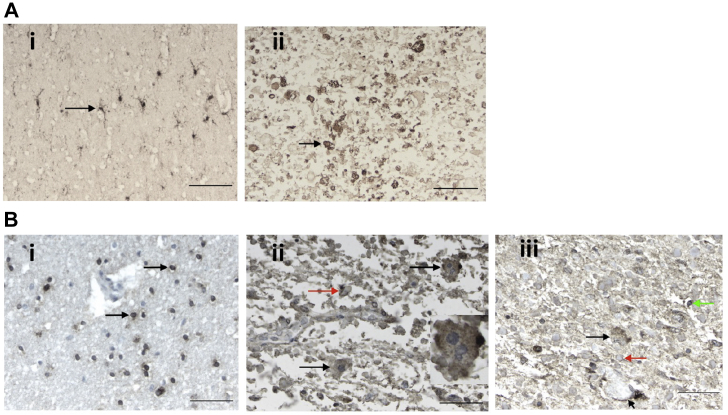
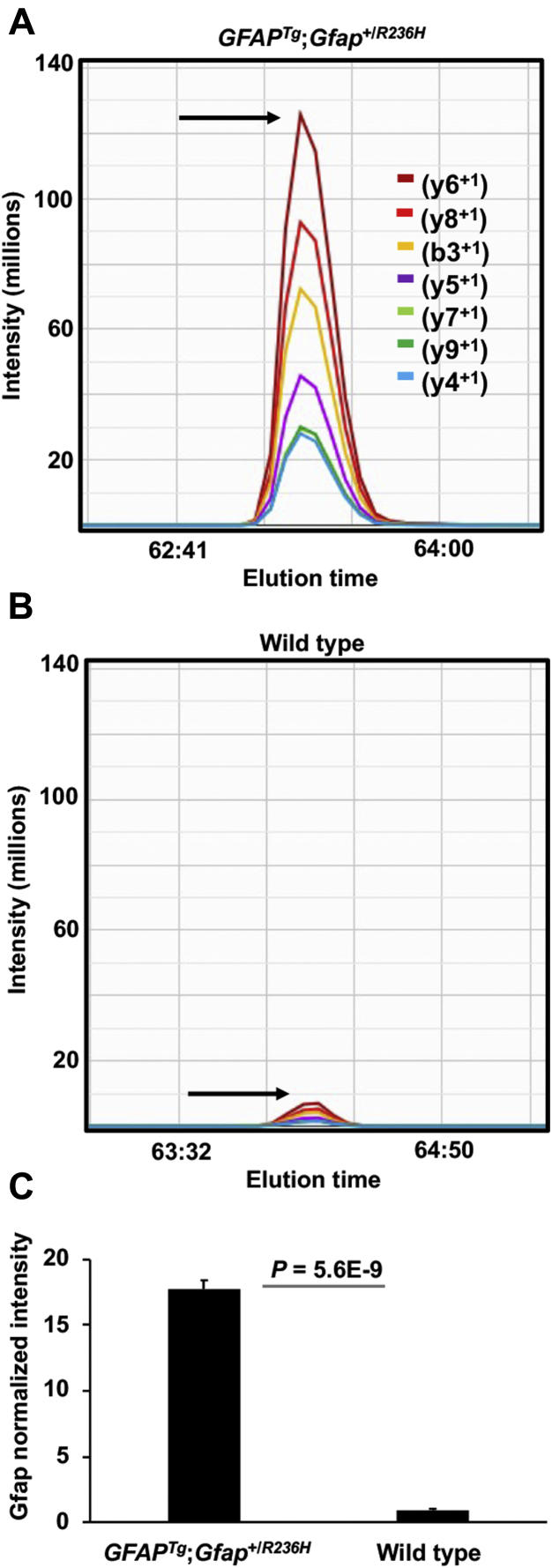
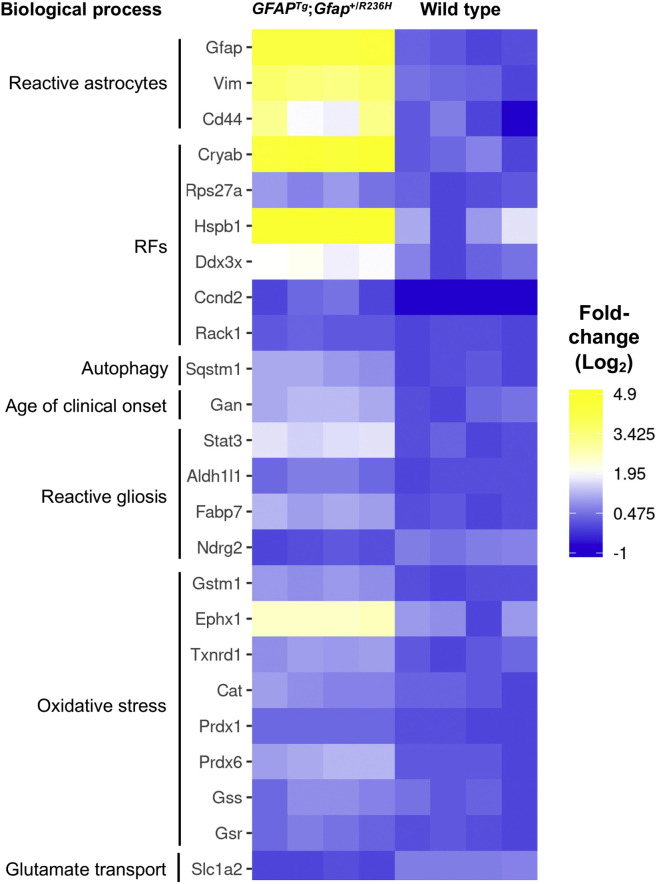
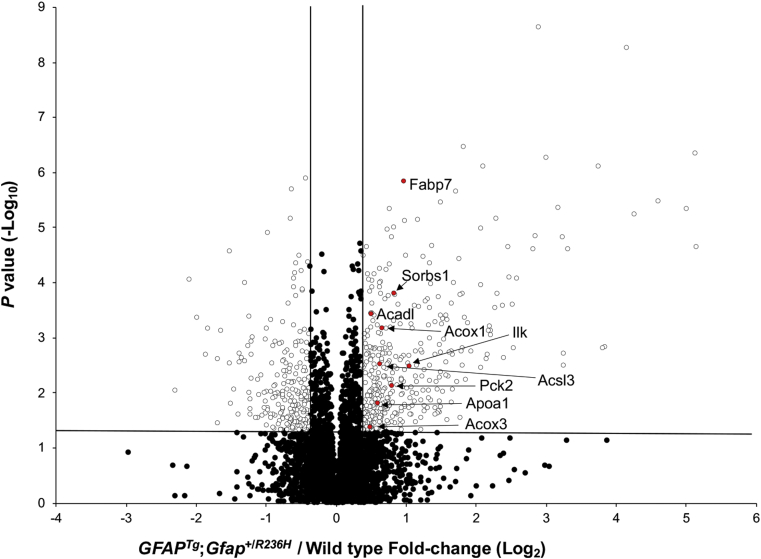
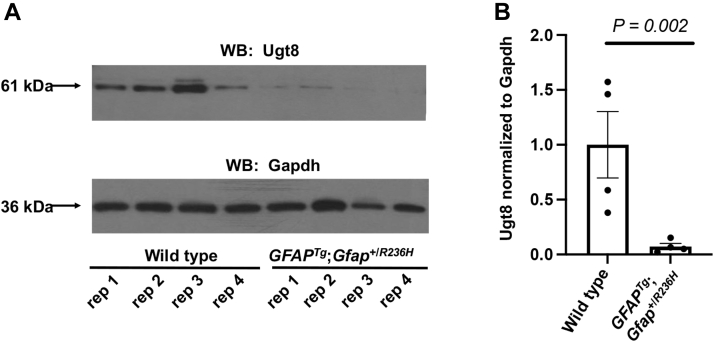
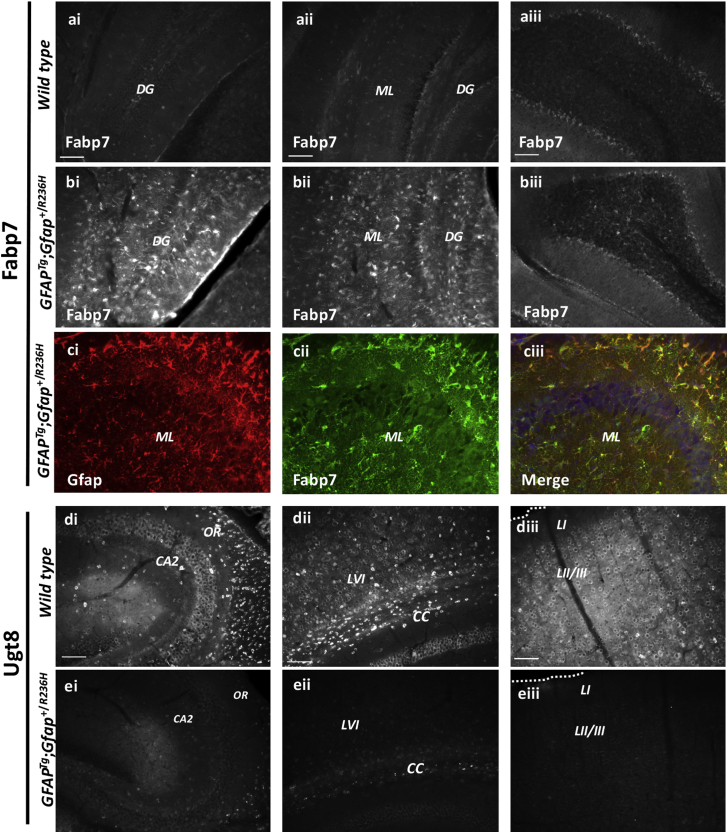
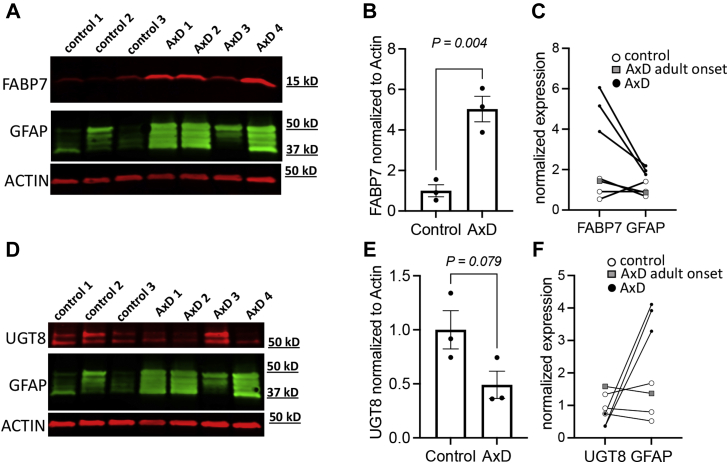
Alexander disease (AxD) is a rare and fatal neurodegenerative disorder caused by mutations in the gene encoding glial fibrillary acidic protein (GFAP). In this report, a mouse model of AxD (GFAP;Gfap) was analyzed that contains a heterozygous R236H point mutation in murine Gfap as well as a transgene with a GFAP promoter to overexpress human GFAP. Using label-free quantitative proteomic comparisons of brain tissue from GFAP;Gfap versus wild-type mice confirmed upregulation of the glutathione metabolism pathway and indicated proteins were elevated in the peroxisome proliferator-activated receptor (PPAR) signaling pathway, which had not been reported previously in AxD. Relative protein-level differences were confirmed by a targeted proteomics assay, including proteins related to astrocytes and oligodendrocytes. Of particular interest was the decreased level of the oligodendrocyte protein, 2-hydroxyacylsphingosine 1-beta-galactosyltransferase (Ugt8), since Ugt8-deficient mice exhibit a phenotype similar to GFAP;Gfap mice (e.g., tremors, ataxia, hind-limb paralysis). In addition, decreased levels of myelin-associated proteins were found in the GFAP;Gfap mice, consistent with the role of Ugt8 in myelin synthesis. Fabp7 upregulation in GFAP;Gfap mice was also selected for further investigation due to its uncharacterized association to AxD, critical function in astrocyte proliferation, and functional ability to inhibit the anti-inflammatory PPAR signaling pathway in models of amyotrophic lateral sclerosis (ALS). Within Gfap astrocytes, Fabp7 was markedly increased in the hippocampus, a brain region subjected to extensive pathology and chronic reactive gliosis in GFAP;Gfap mice. Last, to determine whether the findings in GFAP;Gfap mice are present in the human condition, AxD patient and control samples were analyzed by Western blot, which indicated that Type I AxD patients have a significant fourfold upregulation of FABP7. However, immunohistochemistry analysis showed that UGT8 accumulates in AxD patient subpial brain regions where abundant amounts of Rosenthal fibers are located, which was not observed in the GFAP;Gfap mice.
亚历山大病(AxD)是一种罕见且致命的神经退行性疾病,由编码神经胶质纤维酸性蛋白(GFAP)的基因突变引起。在本报告中,分析了一种 AxD 的小鼠模型(GFAP; Gfap),该模型在小鼠 Gfap 中含有杂合 R236H 点突变,以及一个带有 GFAP 启动子的转基因,以过表达人 GFAP。使用来自 GFAP; Gfap 与野生型小鼠的脑组织的无标记定量蛋白质组学比较证实了谷胱甘肽代谢途径的上调,并表明蛋白质在过氧化物酶体增殖物激活受体(PPAR)信号通路中升高,这在以前的 AxD 中尚未报道过。通过靶向蛋白质组学测定法证实了相对蛋白质水平的差异,包括与星形胶质细胞和少突胶质细胞相关的蛋白质。特别有趣的是少突胶质细胞蛋白 2-羟基酰基鞘氨醇 1-β-半乳糖基转移酶(Ugt8)的水平降低,因为 Ugt8 缺陷型小鼠表现出与 GFAP; Gfap 小鼠相似的表型(例如,震颤,共济失调,后肢瘫痪)。此外,在 GFAP; Gfap 小鼠中发现髓鞘相关蛋白水平降低,这与 Ugt8 在髓鞘合成中的作用一致。由于 Fabp7 在星形胶质细胞增殖中的重要作用及其在肌萎缩侧索硬化症(ALS)模型中抑制抗炎性 PPAR 信号通路的功能能力,GFAP; Gfap 小鼠中 Fabp7 的上调也被选择进行进一步研究。在 Gfap 星形胶质细胞中,Fabp7 在海马体中显著增加,海马体是 GFAP; Gfap 小鼠中广泛发生病理学和慢性反应性神经胶质增生的脑区。最后,为了确定 GFAP; Gfap 小鼠中的发现是否存在于人类疾病中,通过 Western blot 分析了 AxD 患者和对照样本,这表明 I 型 AxD 患者的 FABP7 显著上调了四倍。但是,免疫组织化学分析表明,UGT8 在 AxD 患者软脑膜脑区积聚,在那里大量存在 Rosenthal 纤维,而在 GFAP; Gfap 小鼠中未观察到。