Kovács Péter, Tran Fabien, Hanbury Allan, Madsen Georg K H
Institute of Materials Chemistry, Technical University of Vienna, Getreidemarkt 9/165-TC, A-1060 Vienna, Austria.
Institute for Information Systems Engineering, Technical University of Vienna, Favoritenstrasse 9-11/194, A-1040 Vienna, Austria.
J Chem Theory Comput. 2022 Jan 11;18(1):441-447. doi: 10.1021/acs.jctc.1c00536. Epub 2021 Dec 17.
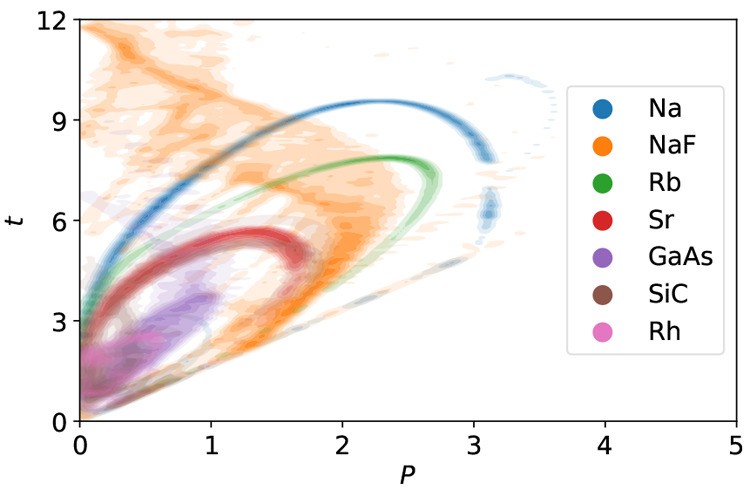
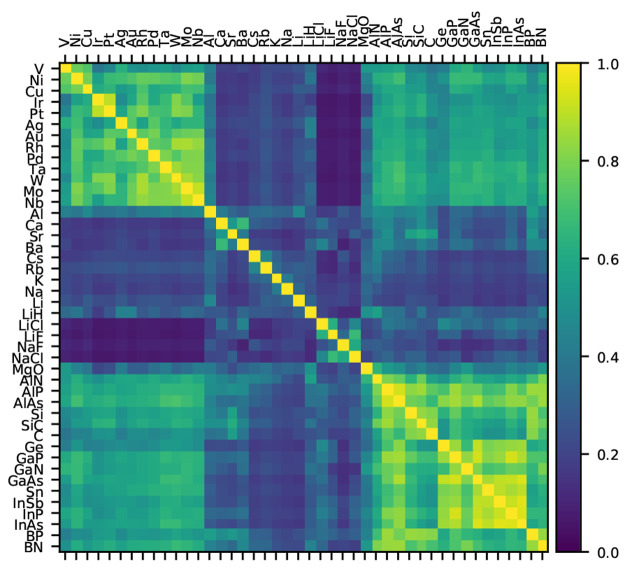
Benchmarking DFT functionals is complicated since the results highly depend on which properties and materials were used in the process. Unwanted biases can be introduced if a data set contains too many examples of very similar materials. We show that a clustering based on the distribution of density gradient and kinetic energy density is able to identify groups of chemically distinct solids. We then propose a method to create smaller data sets or rebalance existing data sets in a way that no region of the meta-GGA descriptor space is overrepresented, yet the new data set reproduces average errors of the original set as closely as possible. We apply the method to an existing set of 44 inorganic solids and suggest a representative set of seven solids. The representative sets generated with this method can be used to make more general benchmarks or to train new functionals.
对密度泛函理论(DFT)泛函进行基准测试很复杂,因为结果高度依赖于过程中使用的性质和材料。如果数据集包含太多非常相似材料的示例,可能会引入不必要的偏差。我们表明,基于密度梯度和动能密度分布的聚类能够识别化学性质不同的固体组。然后,我们提出了一种方法来创建更小的数据集或以某种方式重新平衡现有数据集,使得元广义梯度近似(meta-GGA)描述符空间的任何区域都不会被过度代表,同时新数据集尽可能紧密地重现原始数据集的平均误差。我们将该方法应用于现有的44种无机固体数据集,并提出了一个由七种固体组成的代表性数据集。用这种方法生成的代表性数据集可用于进行更通用的基准测试或训练新的泛函。