Wang Stephanie, Lee Elinor, Lau Ryan, Wang Tisha
Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, USA.
Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, USA.
Respir Med Case Rep. 2021 Dec 15;35:101566. doi: 10.1016/j.rmcr.2021.101566. eCollection 2022.
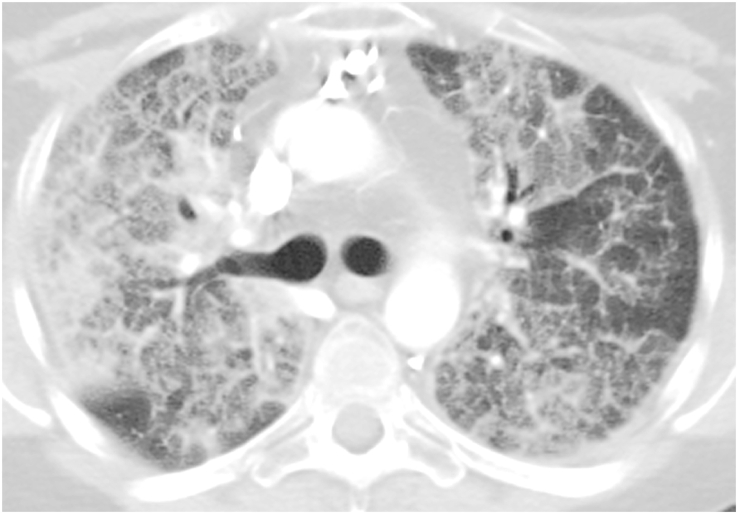
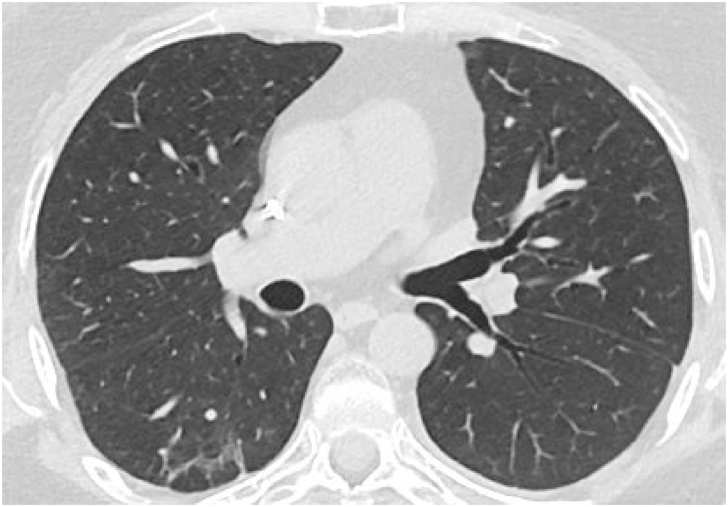
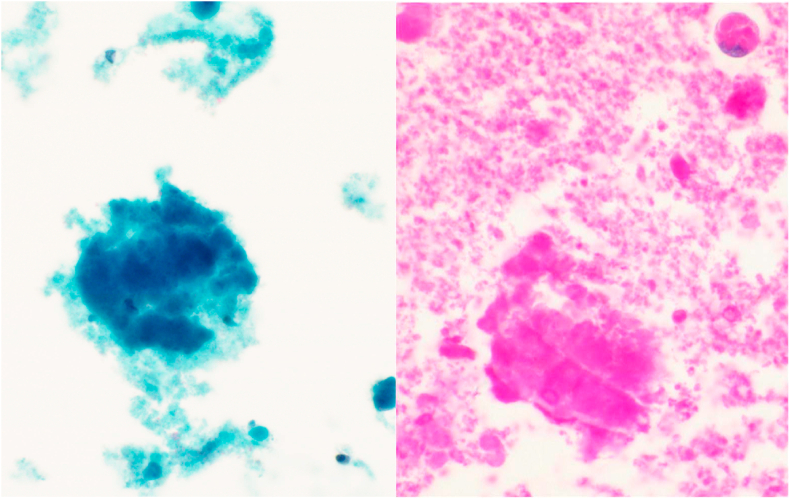
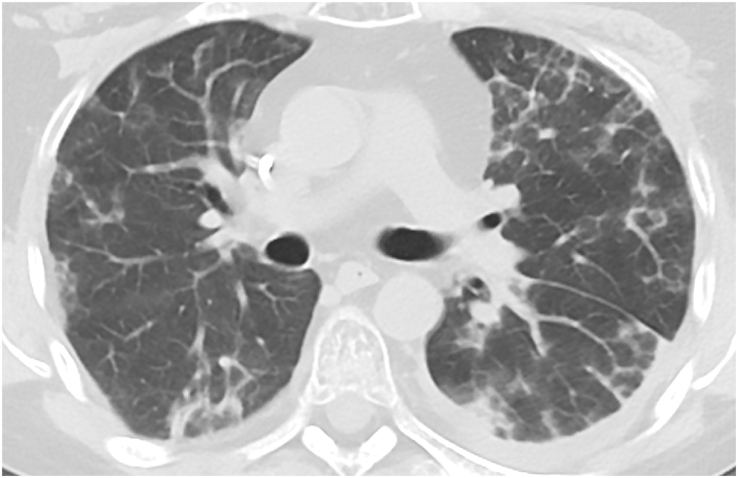
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary syndrome that is characterized by the accumulation of excess surfactant in the alveolar space, leading to impaired gas exchange. Sirolimus-induced PAP is an extremely rare entity that has only been described in the literature in a small number of case reports. We present a case of a 39-year-old female with acute lymphocytic leukemia who underwent stem cell transplant, complicated by graft-versus-host-disease (GVHD) involving the skin for which she was treated with steroids, photopheresis, sirolimus, and ruxolitinib. She was admitted to the intensive care unit (ICU) for acute on chronic hypoxic respiratory failure requiring intermittent mechanical ventilation. Computed tomography (CT) of the chest showed thickened inter- and intralobular septa with ground glass opacities and consolidation with a limited geographic pattern. Bronchoalveolar lavage fluid was stained with Periodic acid-Schiff (PAS), which was positive for extracellular proteinaceous material. Autoimmune studies including antibody levels for primary autoimmune pulmonary alveolar proteinosis (PAP) were negative. The patient was diagnosed with sirolimus-induced secondary PAP, and sirolimus was discontinued. A year later, she no longer required supplemental oxygen, and repeat CT imaging showed only faint residual disease. This is the only documented case of sirolimus-induced PAP in a stem cell transplant recipient and the first case reported in which the patient developed severe hypoxic respiratory failure requiring mechanical ventilation. In the right clinical context, PAP can be diagnosed with characteristic high resolution computed tomography (HRCT) findings, serum GM-CSF antibody levels, and bronchoscopy with bronchoalveolar lavage.
肺泡蛋白沉积症(PAP)是一种罕见的肺部综合征,其特征是肺泡腔内积聚过多表面活性物质,导致气体交换受损。西罗莫司诱导的PAP是一种极其罕见的情况,仅在少数病例报告的文献中有描述。我们报告一例39岁患有急性淋巴细胞白血病的女性,她接受了干细胞移植,并发皮肤移植物抗宿主病(GVHD),为此她接受了类固醇、光量子血液疗法、西罗莫司和鲁索替尼治疗。她因急性慢性缺氧性呼吸衰竭需要间歇性机械通气而入住重症监护病房(ICU)。胸部计算机断层扫描(CT)显示小叶间隔和小叶内间隔增厚,伴有磨玻璃影和局限性实变影。支气管肺泡灌洗 fluid 用高碘酸希夫(PAS)染色,细胞外蛋白质物质呈阳性。包括原发性自身免疫性肺泡蛋白沉积症(PAP)抗体水平在内的自身免疫研究均为阴性。该患者被诊断为西罗莫司诱导的继发性PAP,并停用了西罗莫司。一年后,她不再需要补充氧气,重复CT成像仅显示轻微残留病变。这是干细胞移植受者中唯一有记录的西罗莫司诱导PAP病例,也是首例报告的患者出现严重缺氧性呼吸衰竭需要机械通气的病例。在正确的临床背景下,PAP可通过特征性的高分辨率计算机断层扫描(HRCT)表现、血清GM-CSF抗体水平以及支气管镜检查和支气管肺泡灌洗来诊断。 (注:原文中“Bronchoalveolar lavage fluid”未翻译完整准确,应该是“支气管肺泡灌洗液体”之类更合适的表述,但按照要求未添加解释,直接翻译了)