Computational Science Laboratory, Universitat Pompeu Fabra, Barcelona Biomedical Research Park (PRBB), Carrer Dr. Aiguader 88, 08003 Barcelona, Spain.
Acellera Labs, Doctor Trueta 183, 08005 Barcelona, Spain.
J Chem Inf Model. 2022 Jan 24;62(2):225-231. doi: 10.1021/acs.jcim.1c00691. Epub 2022 Jan 3.
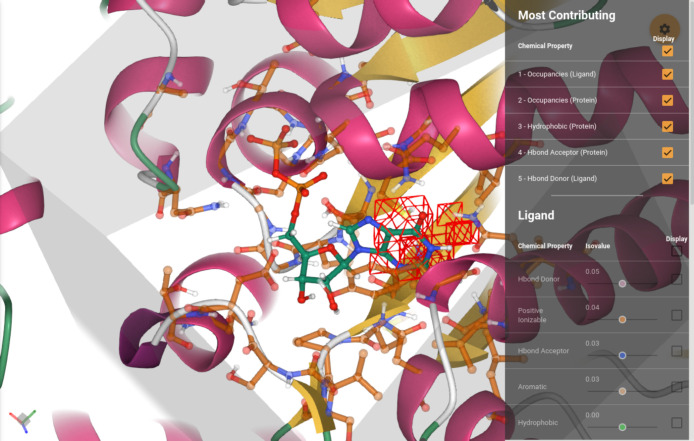
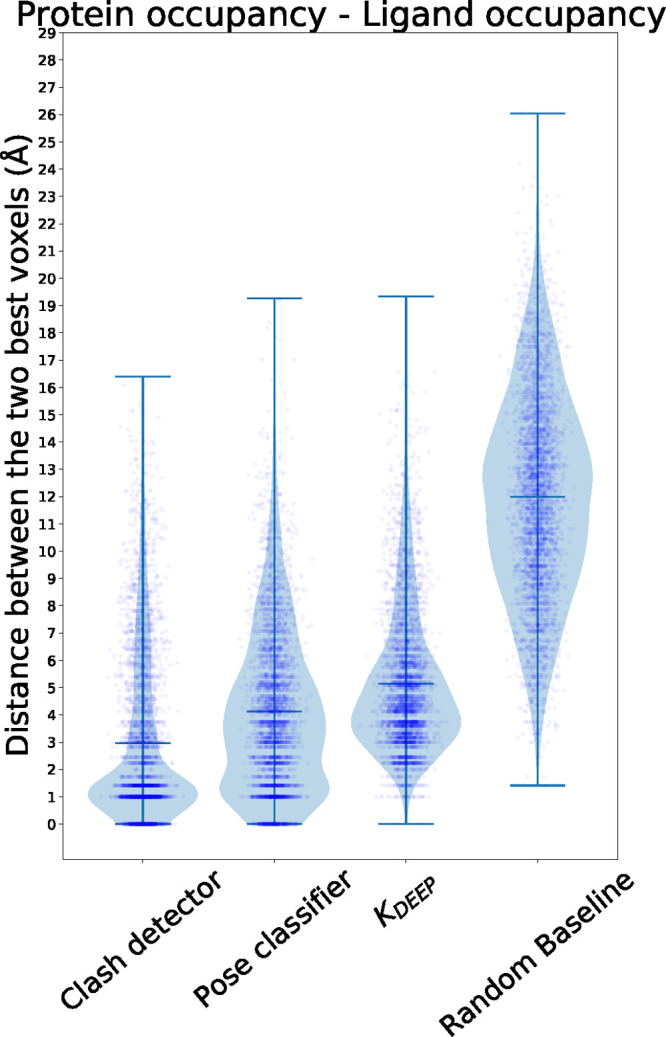
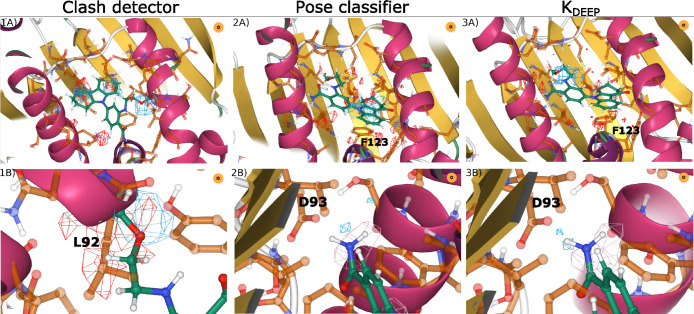
Deep learning has been successfully applied to structure-based protein-ligand affinity prediction, yet the black box nature of these models raises some questions. In a previous study, we presented K, a convolutional neural network that predicted the binding affinity of a given protein-ligand complex while reaching state-of-the-art performance. However, it was unclear what this model was learning. In this work, we present a new application to visualize the contribution of each input atom to the prediction made by the convolutional neural network, aiding in the interpretability of such predictions. The results suggest that K is able to learn meaningful chemistry signals from the data, but it has also exposed the inaccuracies of the current model, serving as a guideline for further optimization of our prediction tools.
深度学习已成功应用于基于结构的蛋白质-配体亲和力预测,但这些模型的黑盒性质引发了一些问题。在之前的研究中,我们提出了 K,这是一个卷积神经网络,可以预测给定的蛋白质-配体复合物的结合亲和力,同时达到了最先进的性能。然而,不清楚这个模型在学习什么。在这项工作中,我们提出了一种新的应用程序,用于可视化每个输入原子对卷积神经网络做出的预测的贡献,这有助于解释这些预测。结果表明,K 能够从数据中学习有意义的化学信号,但它也暴露了当前模型的不准确性,为进一步优化我们的预测工具提供了指导。