Needleman Center for Neurometabolism and Axonal Therapeutics and Department of Genetics, Washington University School of Medicine in Saint Louis, St. Louis, MO, USA.
Needleman Center for Neurometabolism and Axonal Therapeutics and Department of Developmental Biology, Washington University School of Medicine in Saint Louis, St. Louis, MO, USA.
Mol Neurodegener. 2022 Jan 6;17(1):1. doi: 10.1186/s13024-021-00511-x.
In response to injury, neurons activate a program of organized axon self-destruction initiated by the NAD hydrolase, SARM1. In healthy neurons SARM1 is autoinhibited, but single amino acid changes can abolish autoinhibition leading to constitutively active SARM1 enzymes that promote degeneration when expressed in cultured neurons.
To investigate whether naturally occurring human variants might disrupt SARM1 autoinhibition and potentially contribute to risk for neurodegenerative disease, we assayed the enzymatic activity of all 42 rare SARM1 alleles identified among 8507 amyotrophic lateral sclerosis (ALS) patients and 9671 controls. We then intrathecally injected mice with virus expressing SARM1 constructs to test the capacity of an ALS-associated constitutively active SARM1 variant to promote neurodegeneration in vivo.
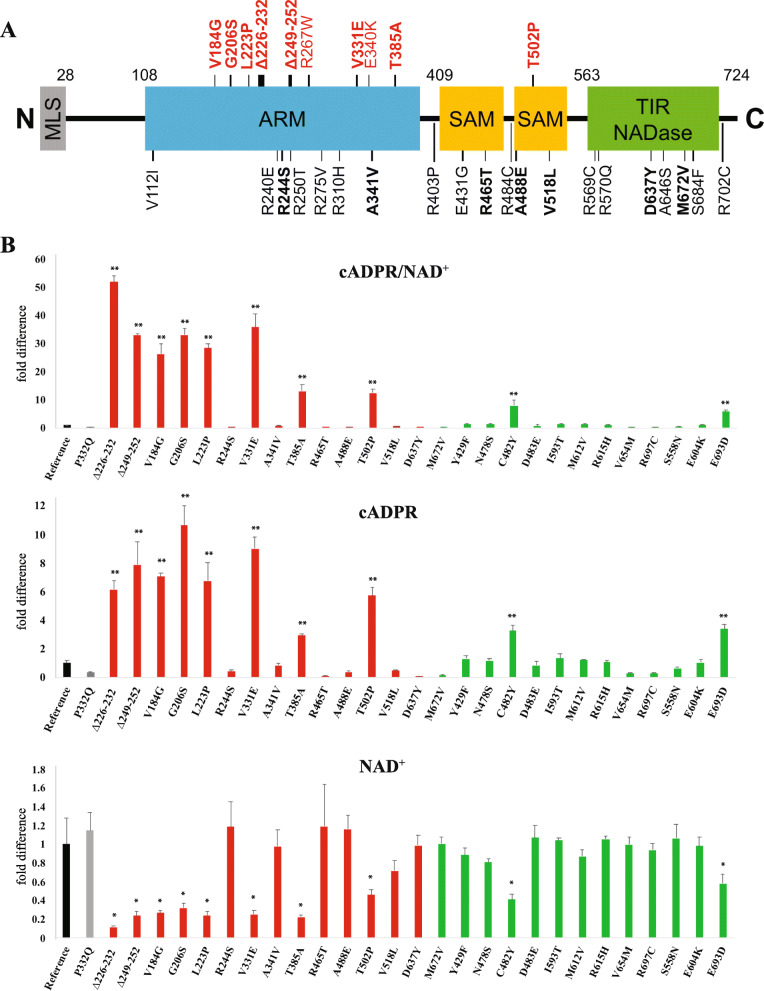
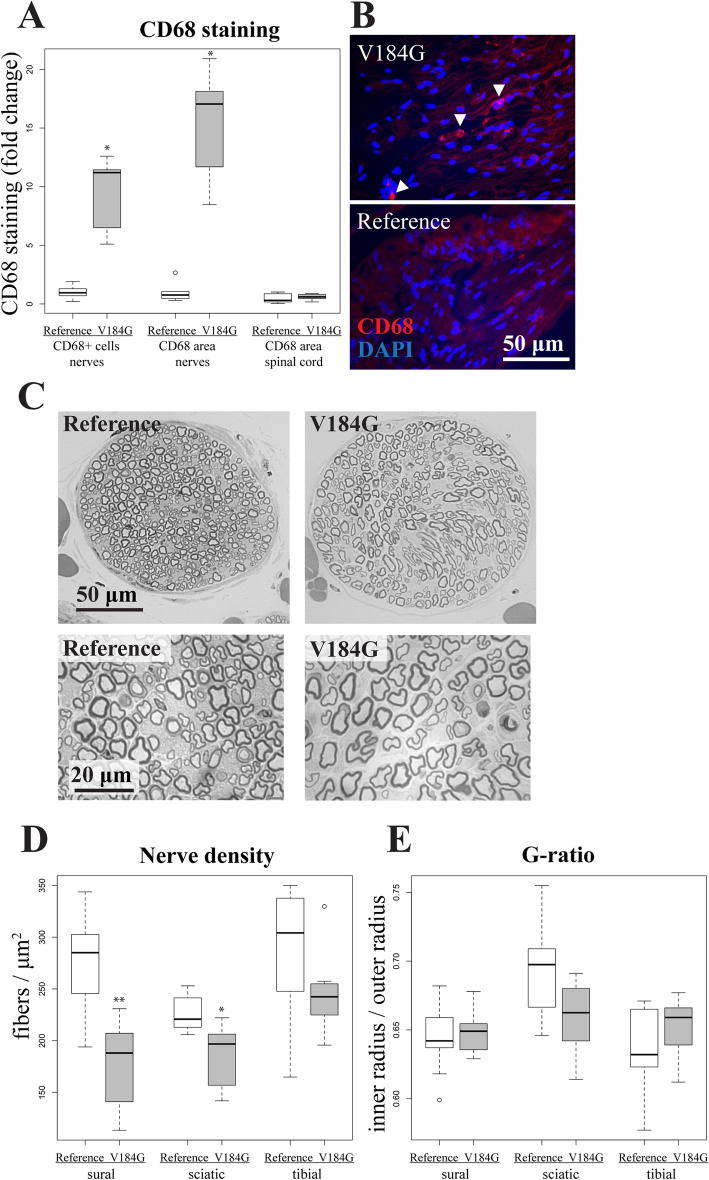
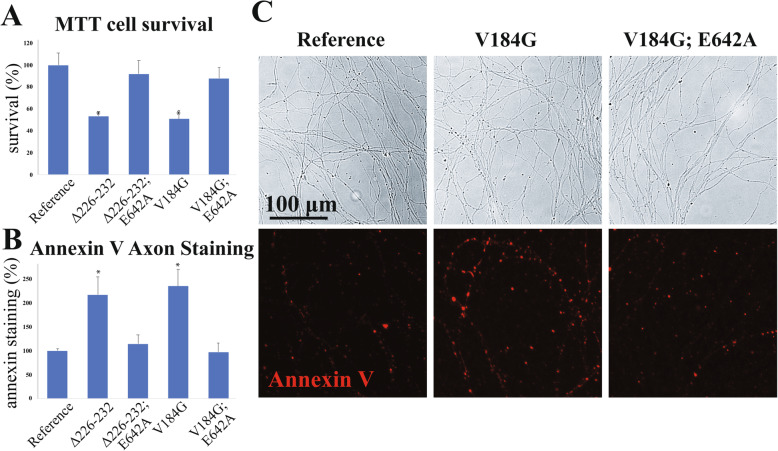
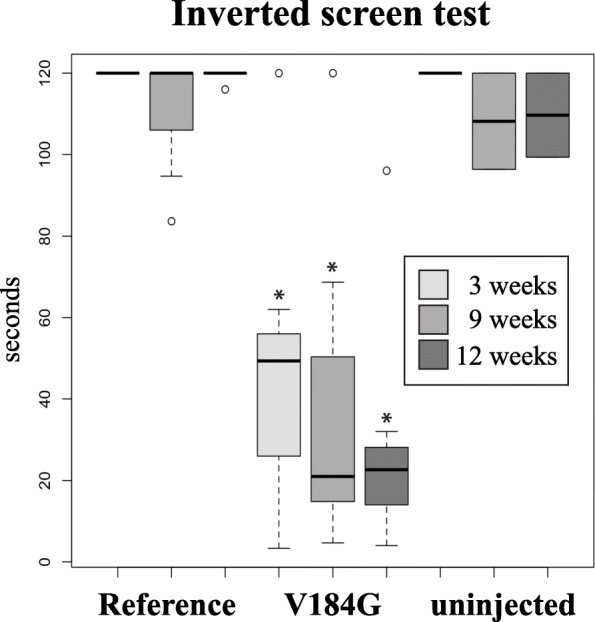
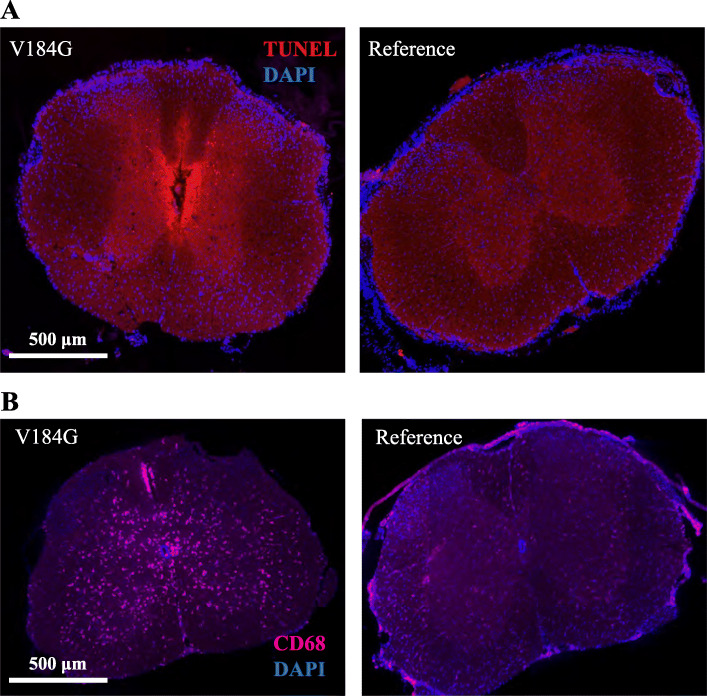
Twelve out of 42 SARM1 missense variants or small in-frame deletions assayed exhibit constitutive NADase activity, including more than half of those that are unique to the ALS patients or that occur in multiple patients. There is a > 5-fold enrichment of constitutively active variants among patients compared to controls. Expression of constitutively active ALS-associated SARM1 alleles in cultured dorsal root ganglion (DRG) neurons is pro-degenerative and cytotoxic. Intrathecal injection of an AAV expressing the common SARM1 reference allele is innocuous to mice, but a construct harboring SARM1, the constitutively active variant found most frequently among the ALS patients, causes axon loss, motor dysfunction, and sustained neuroinflammation.
These results implicate rare hypermorphic SARM1 alleles as candidate genetic risk factors for ALS and other neurodegenerative conditions.
神经元在受到损伤时会激活一个有组织的轴突自我毁灭程序,该程序由 NAD 水解酶 SARM1 引发。在健康神经元中,SARM1 被自身抑制,但单个氨基酸的改变可能会消除自身抑制,导致组成型激活的 SARM1 酶,当在培养的神经元中表达时会促进退化。
为了研究天然存在的人类变体是否可能破坏 SARM1 的自身抑制作用,并可能导致神经退行性疾病的风险,我们检测了在 8507 名肌萎缩侧索硬化症 (ALS) 患者和 9671 名对照者中发现的所有 42 种罕见 SARM1 等位基因的酶活性。然后,我们通过鞘内注射表达 SARM1 构建体的病毒来测试与 ALS 相关的组成型激活的 SARM1 变体在体内促进神经退行性变的能力。
在所检测的 42 种 SARM1 错义变异体或小框内缺失中,有 12 种表现出组成型 NADase 活性,其中包括超过一半的仅存在于 ALS 患者或在多个患者中发生的变异体。与对照组相比,患者中组成型激活变体的富集程度超过 5 倍。在培养的背根神经节 (DRG) 神经元中表达组成型激活的 ALS 相关 SARM1 等位基因具有促退化和细胞毒性作用。鞘内注射表达常见 SARM1 参考等位基因的 AAV 对小鼠无害,但携带 SARM1 的构建体,该变体最常见于 ALS 患者,会导致轴突丢失、运动功能障碍和持续的神经炎症。
这些结果表明,罕见的超型 SARM1 等位基因是 ALS 和其他神经退行性疾病的候选遗传风险因素。