Sun Menghuai, Lin Qian, Wang Chunyang, Xing Jiao, Yan Kunlong, Liu Zhifeng, Jin Yu, Cardona Carol J, Xing Zheng
Medical School and Jiangsu Provincial Key Laboratory of Medicine, Nanjing University, Nanjing, China.
Nanjing Children's Hospital, Nanjing Medical University, Nanjing, China.
Front Microbiol. 2021 Dec 21;12:762869. doi: 10.3389/fmicb.2021.762869. eCollection 2021.
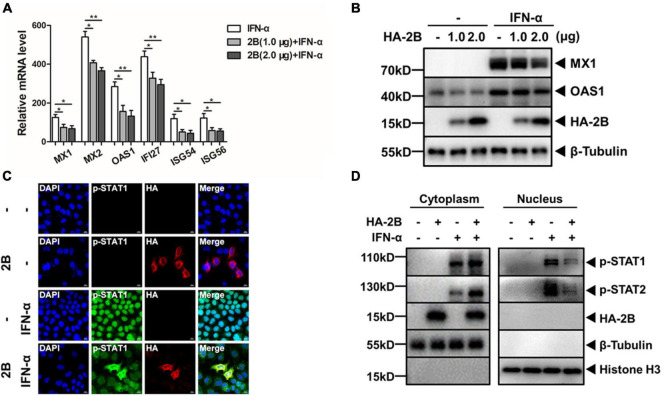
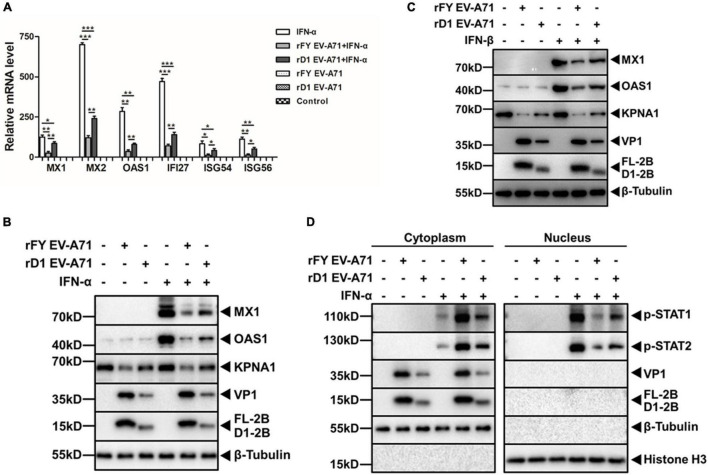
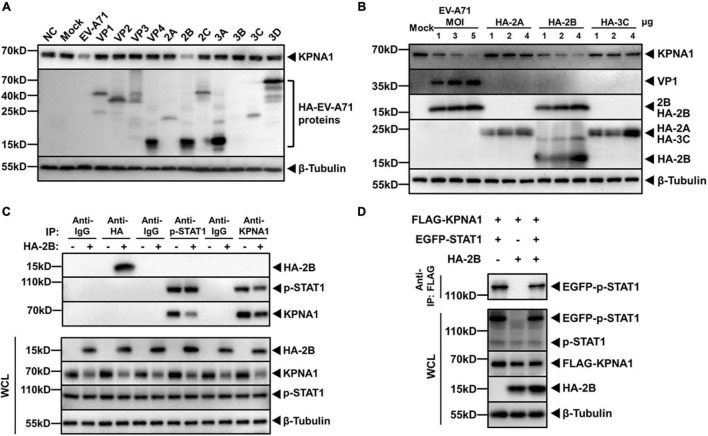
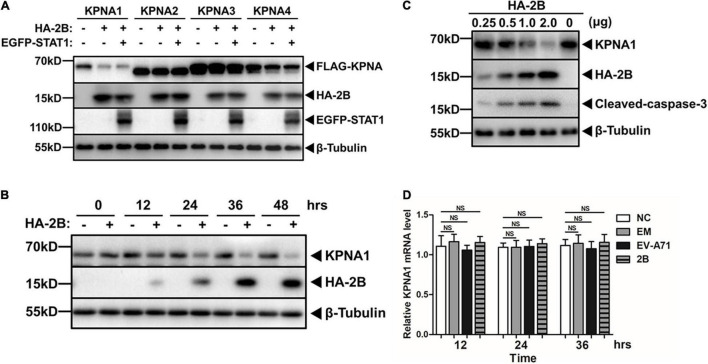
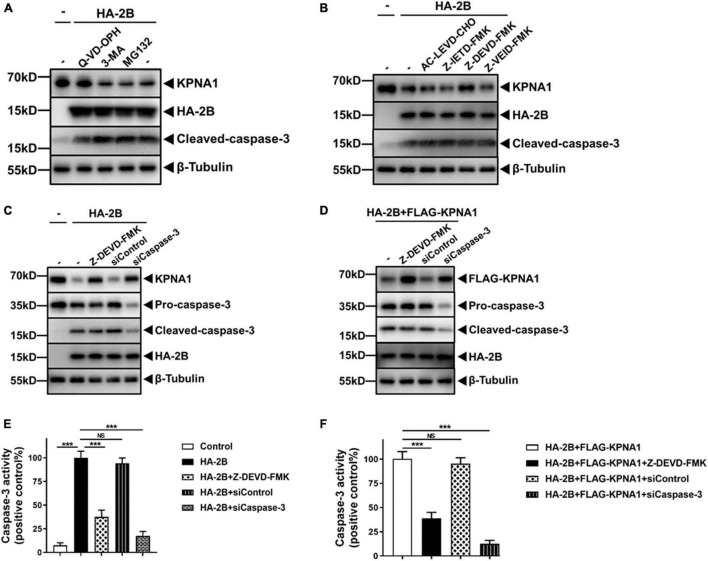
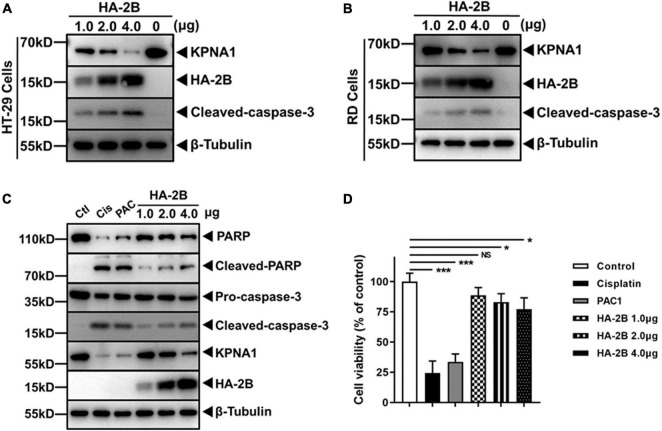
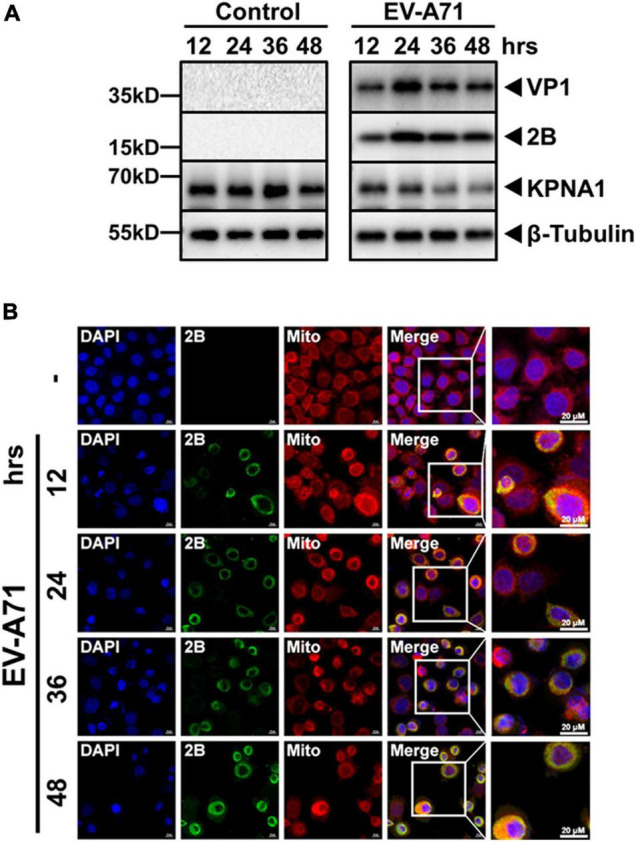
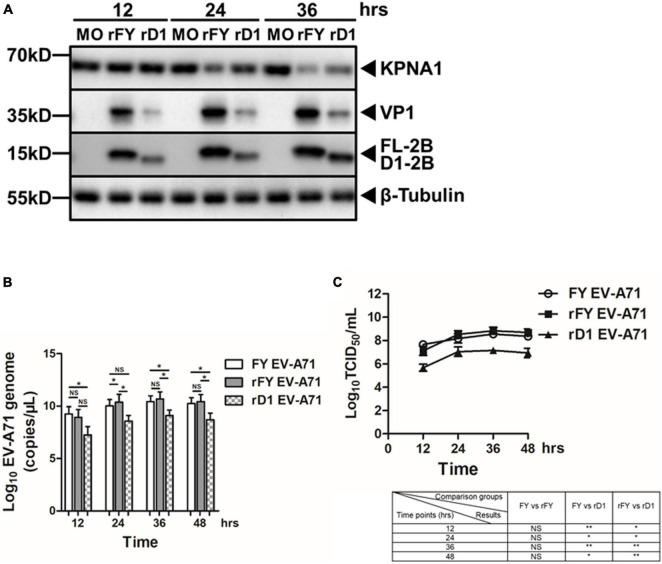
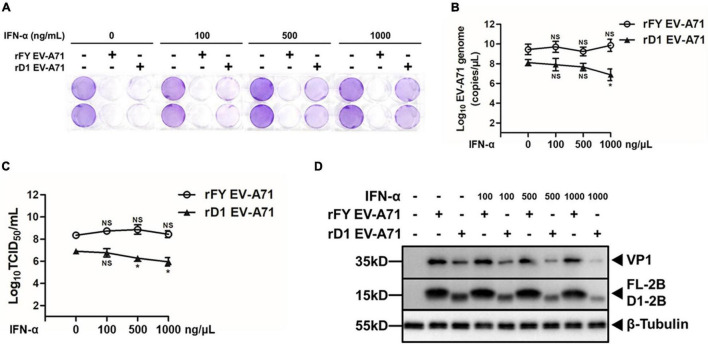
Enterovirus A71 (EV-A71) is a major pathogen that causes the hand, foot, and mouth disease, which could be fatal with neurological complications in children. The underlying mechanism for the severe pathogenicity remains obscure, but impaired or aberrant innate immunity is considered to play a key role in viral pathogenesis. We reported previously that EV-A71 suppressed type I interferon (IFN) responses by inducing degradation of karyopherin-α1 (KPNA1), a component of the p-STAT1/2 complex. In this report, we showed that 2B, a non-structural protein of EV-A71, was critical to the suppression of the IFN-α-induced type I response in infected cells. Among viral proteins, 2B was the only one that was involved in the degradation of KPNA1, which impeded the formation of the p-STAT1/2/KPNA1 complex and blocked the translocation of p-STAT1/2 into the nucleus upon IFN-α stimulation. Degradation of KPNA1 induced by 2B can be inhibited in the cells pre-treated with Z-DEVD-FMK, a caspase-3 inhibitor, or siRNA targeting caspase-3, indicating that 2B-induced degradation of KPNA1 was caspase-3 dependent. The mechanism by which 2B functioned in the dysregulation of the IFN signaling was analyzed and a putative hydrophilic domain (H1) in the N-terminus of 2B was characterized to be critical for the release of cytochrome c into the cytosol for the activation of pro-caspase-3. We generated an EV-A71 infectious clone (rD1), which was deficient of the H1 domain. In rD1-infected cells, degradation of KPNA1 was relieved and the infected cells were more sensitive to IFN-α, leading to decreased viral replication, in comparison to the cells infected with the virus carrying a full length 2B. Our findings demonstrate that EV-A71 2B protein plays an important role in dysregulating JAK-STAT signaling through its involvement in promoting caspase-3 dependent degradation of KPNA1, which represents a novel strategy employed by EV-A71 to evade host antiviral innate immunity.
肠道病毒A71(EV - A71)是导致手足口病的主要病原体,该病在儿童中可能引发致命的神经并发症。其严重致病性的潜在机制仍不清楚,但受损或异常的固有免疫被认为在病毒发病机制中起关键作用。我们之前报道过,EV - A71通过诱导核转运蛋白α1(KPNA1)降解来抑制I型干扰素(IFN)反应,KPNA1是p - STAT1/2复合物的一个组成部分。在本报告中,我们表明EV - A71的非结构蛋白2B对于抑制感染细胞中IFN - α诱导的I型反应至关重要。在病毒蛋白中,2B是唯一参与KPNA1降解的蛋白,这阻碍了p - STAT1/2/KPNA1复合物的形成,并在IFN - α刺激时阻止p - STAT1/2转运到细胞核中。用caspase - 3抑制剂Z - DEVD - FMK或靶向caspase - 3的小干扰RNA(siRNA)预处理细胞后,可抑制2B诱导的KPNA1降解,这表明2B诱导的KPNA1降解依赖于caspase - 3。分析了2B在IFN信号失调中发挥作用的机制,并确定2B N端一个假定的亲水区(H1)对于细胞色素c释放到细胞质中以激活前体caspase - 3至关重要。我们构建了一个缺失H1结构域的EV - A71感染性克隆(rD1)。与感染携带全长2B病毒的细胞相比,在rD1感染的细胞中,KPNA1的降解得到缓解,感染细胞对IFN - α更敏感,导致病毒复制减少。我们的研究结果表明,EV - A71 2B蛋白通过促进caspase - 3依赖的KPNA1降解参与调节JAK - STAT信号,这代表了EV - A71逃避宿主抗病毒固有免疫的一种新策略。