Department of Internal Medicine and Paediatrics, Ghent University, Ghent B-9000, Belgium.
Center for Inflammation Research, VIB, Ghent B-9000, Belgium.
Proc Natl Acad Sci U S A. 2022 Jan 11;119(2). doi: 10.1073/pnas.2116415119.
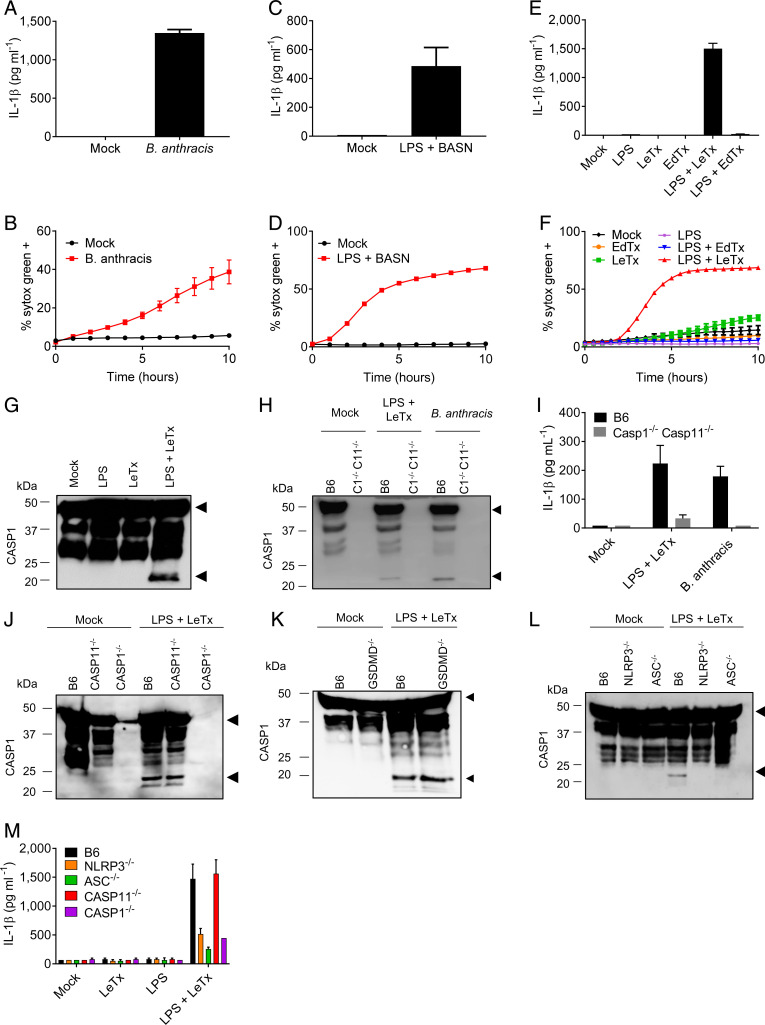
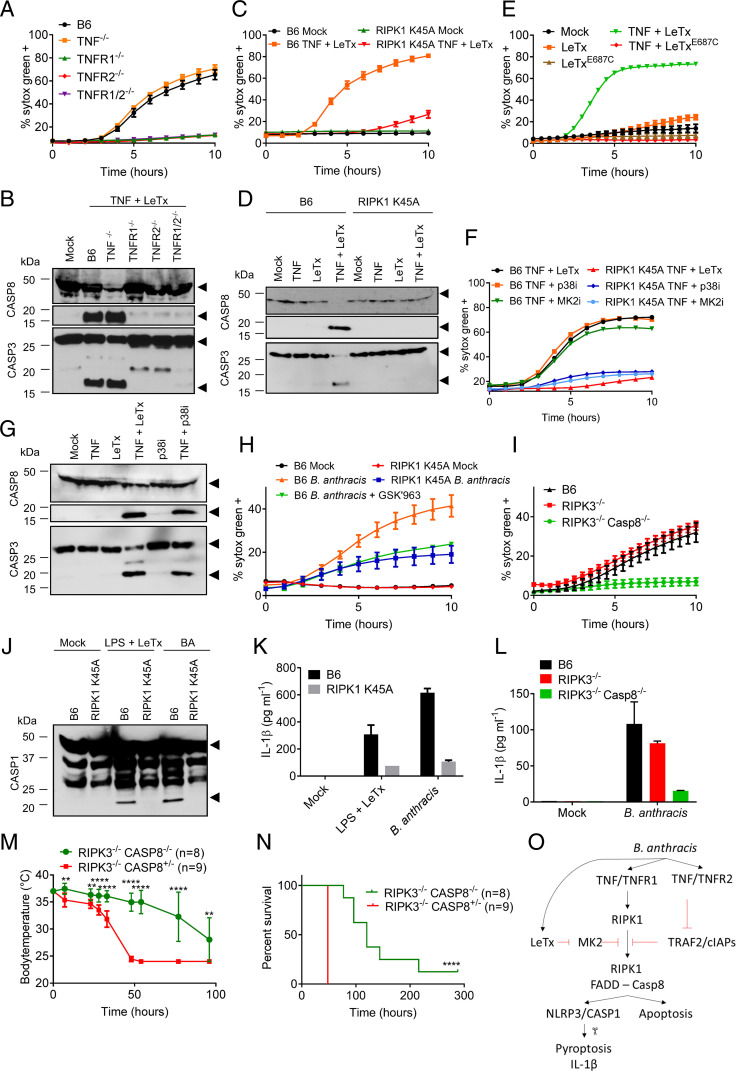
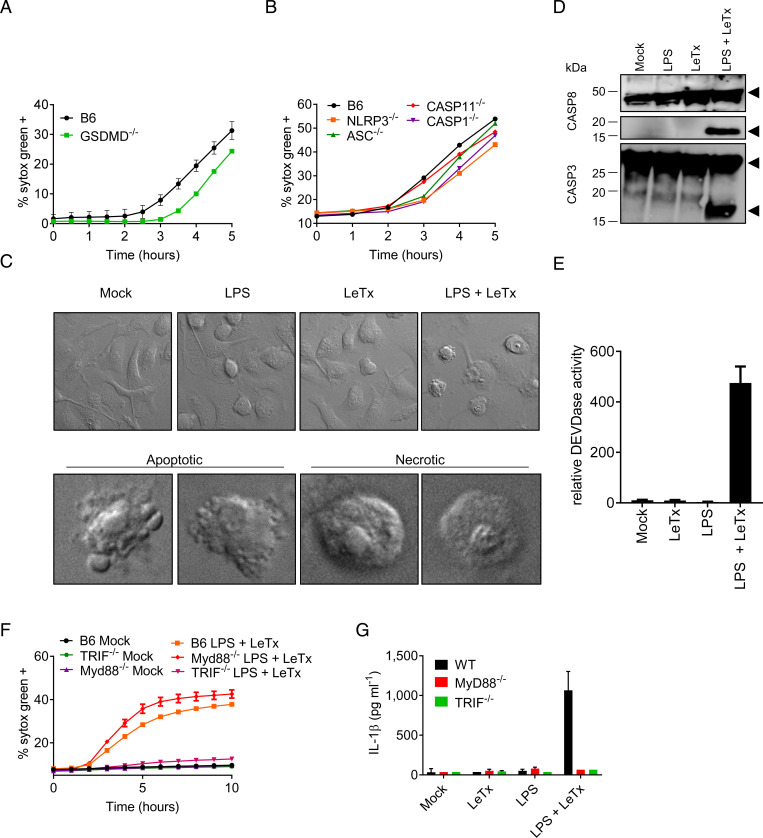
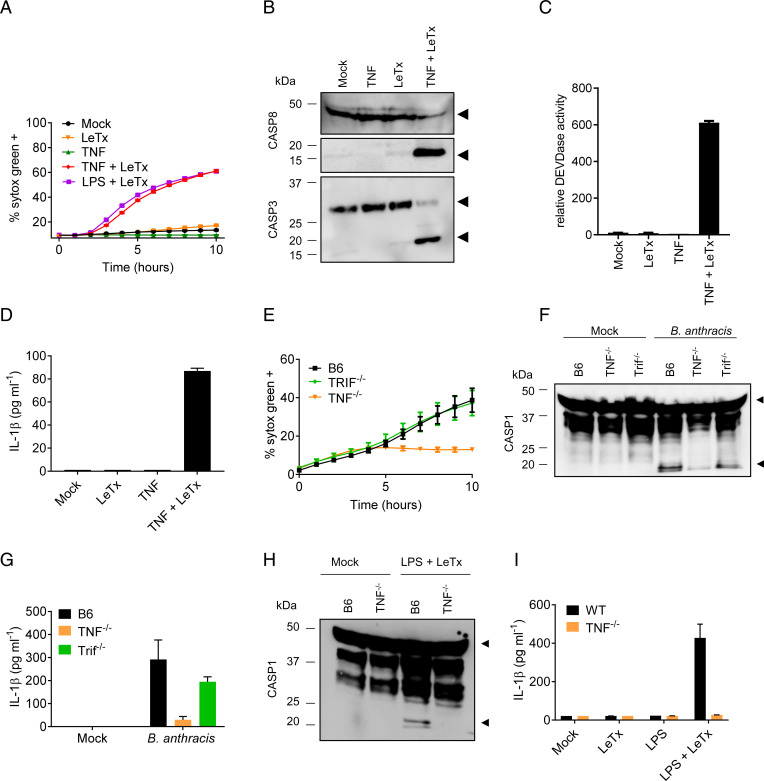
Lethal toxin (LeTx)-mediated killing of myeloid cells is essential for the causative agent of anthrax, to establish systemic infection and induce lethal anthrax. The "LeTx-sensitive" NLRP1b inflammasome of BALB/c and 129S macrophages swiftly responds to LeTx intoxication with pyroptosis and secretion of interleukin (IL)-1β. However, human NLRP1 is nonresponsive to LeTx, prompting us to investigate host-pathogen interactions in C57BL/6J (B6) macrophages and mice that also lack a LeTx-sensitive allele. Unexpectedly, we found that LeTx intoxication and live infection of B6 macrophages elicited robust secretion of IL-1β, which critically relied on the NLRP3 inflammasome. TNF signaling through both TNF receptor 1 (TNF-R1) and TNF-R2 were required for induced NLRP3 inflammasome activation, which was further controlled by RIPK1 kinase activity and LeTx-mediated proteolytic inactivation of MAP kinase signaling. In addition to activating the NLRP3 inflammasome, LeTx-induced MAPKK inactivation and TNF production sensitized -infected macrophages to robust RIPK1- and caspase-8-dependent apoptosis. In agreement, purified LeTx triggered RIPK1 kinase activity- and caspase-8-dependent apoptosis only in macrophages primed with TNF or following engagement of TRIF-dependent Toll-like receptors. Consistently, genetic and pharmacological inhibition of RIPK1 inhibited NLRP3 inflammasome activation and apoptosis of LeTx-intoxicated and -infected macrophages. Caspase-8/RIPK3-deficient mice were significantly protected from -induced lethality, demonstrating the in vivo pathophysiological relevance of this cytotoxic mechanism. Collectively, these results establish TNF- and RIPK1 kinase activity-dependent NLRP3 inflammasome activation and macrophage apoptosis as key host-pathogen mechanisms in lethal anthrax.
致死毒素 (LeTx) 介导的髓样细胞杀伤对于炭疽病的病原体至关重要,它可导致全身感染并引发致命性炭疽病。BALB/c 和 129S 巨噬细胞中的“LeTx 敏感”NLRP1b 炎性小体对 LeTx 中毒迅速做出反应,导致细胞发生细胞焦亡并分泌白细胞介素 (IL)-1β。然而,人类 NLRP1 对 LeTx 无反应,促使我们研究 C57BL/6J(B6)巨噬细胞和缺乏 LeTx 敏感等位基因的小鼠中的宿主-病原体相互作用。出乎意料的是,我们发现 B6 巨噬细胞在受到 LeTx 中毒和活细菌感染时会强烈分泌 IL-1β,这严重依赖于 NLRP3 炎性小体。TNF 通过 TNF 受体 1(TNF-R1)和 TNF-R2 的信号转导对于诱导的 NLRP3 炎性小体激活是必需的,而该激活过程进一步受到 RIPK1 激酶活性和 LeTx 介导的 MAP 激酶信号通路的蛋白水解失活的控制。除了激活 NLRP3 炎性小体之外,LeTx 诱导的 MAPKK 失活和 TNF 产生使感染的巨噬细胞对 RIPK1 和 caspase-8 依赖性细胞凋亡变得敏感。与此一致,纯化的 LeTx 仅在 TNF 预处理的巨噬细胞或在 TLR 依赖于 TRIF 的情况下引发,才能触发 RIPK1 激酶活性和 caspase-8 依赖性细胞凋亡。同样,RIPK1 的遗传和药理学抑制作用抑制了 LeTx 中毒和感染的巨噬细胞中 NLRP3 炎性小体的激活和细胞凋亡。 caspase-8/RIPK3 缺陷型小鼠从炭疽诱导的致死性中得到了显著保护,这证明了这种细胞毒性机制在体内的病理生理学相关性。总而言之,这些结果确立了 TNF 和 RIPK1 激酶活性依赖性 NLRP3 炎性小体激活和巨噬细胞凋亡作为炭疽病致死的关键宿主-病原体机制。