Dohál Matúš, Dvořáková Věra, Šperková Miluše, Porvazník Igor, Cabibbe Andrea Maurizio, Trovato Alberto, Spitaleri Andrea, Rasmussen Erik Michael, Pršo Kristián, Škereňová Mária, Cirillo Daniela Maria, Solovič Ivan, Mokrý Juraj
Department of Pharmacology and Biomedical Center Martin, Jessenius Faculty of Medicine, Comenius University, Slovakia.
National Reference Laboratory for Mycobacteria, National Institute of Public Health, Praha, Czech Republic.
J Clin Tuberc Other Mycobact Dis. 2021 Dec 20;26:100292. doi: 10.1016/j.jctube.2021.100292. eCollection 2022 Feb.
The resistance of () to antituberculosis drugs poses a major threat to global public health. Whole genome sequencing (WGS) is an increasingly preferred method in the diagnostics and monitoring of the transmission dynamics of resistant forms of tuberculosis (TB). The aim of the study was to, for the first time, use the sequencing-based analysis to study the transmission and resistance patterns of a systematic and recent collection of extensively drug resistant (XDR) and multidrug resistant tuberculosis (MDR-TB) isolates and to expand our knowledge about drug resistant (DR) TB epidemiological dynamics in Slovakia.
A total of 495 patients with pulmonary TB, who were referred to National Reference Laboratory for Mycobacteriology (Vyšné Hágy, Slovakia) in the years 2018-2019, were studied. Out of the total of 495 patients, 4 XDR-TB (0.8%) and 8 (1.6%) MDR-TB isolates were identified by conventional drug susceptibility testing on Löwenstein-Jensen solid medium and subjected to whole genome sequencing. Sequencing data were evaluated for molecular-epidemiological analysis and identification of resistance patterns.
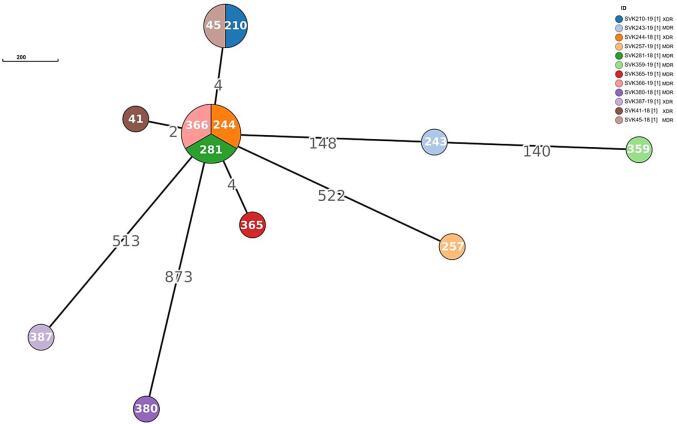
Phylogenetic and cluster analysis showed extensive recent transmission events and the predominance of Euro-American lineage 4.7 in Slovakia. However, phylogenetic analysis revealed the circulation of several lineages that originally occurred in Eastern European countries. Resistance patterns for first- and second-line antituberculosis drugs characterized by whole genome sequencing were in high concordance with the results of phenotypic drug susceptibility testing.
Forty percent of at least MDR-TB isolates were not genetically linked, indicating that appropriate measures should be taken to monitor and prevent the spread of drug-resistant tuberculosis within the country as well as in other regions.
()对抗结核药物的耐药性对全球公共卫生构成重大威胁。全基因组测序(WGS)在耐药结核病(TB)传播动态的诊断和监测中越来越受到青睐。本研究的目的是首次使用基于测序的分析方法,研究近期系统性收集的广泛耐药(XDR)和耐多药结核病(MDR-TB)分离株的传播和耐药模式,并扩展我们对斯洛伐克耐药结核病(DR-TB)流行病学动态的认识。
对2018 - 2019年转诊至国家分枝杆菌病参考实验室(斯洛伐克维什涅哈伊)的495例肺结核患者进行了研究。在这495例患者中,通过在洛温斯坦 - 詹森固体培养基上进行的传统药敏试验,鉴定出4株XDR-TB(0.8%)和8株(1.6%)MDR-TB分离株,并对其进行全基因组测序。对测序数据进行评估,以进行分子流行病学分析和耐药模式鉴定。
系统发育和聚类分析显示,近期存在广泛的传播事件,且斯洛伐克以欧美谱系4.7为主。然而,系统发育分析揭示了几个最初出现在东欧国家的谱系的传播情况。通过全基因组测序确定的一线和二线抗结核药物的耐药模式与表型药敏试验结果高度一致。
至少40%的MDR-TB分离株没有遗传关联,这表明应采取适当措施监测和预防耐药结核病在该国以及其他地区的传播。