Bermejo-Guerrero Laura, de Fuenmayor-Fernández de la Hoz Carlos Pablo, Serrano-Lorenzo Pablo, Blázquez-Encinar Alberto, Gutiérrez-Gutiérrez Gerardo, Martínez-Vicente Laura, Galán-Dávila Lucía, García-García Jorge, Arenas Joaquín, Muelas Nuria, Hernández-Laín Aurelio, Domínguez-González Cristina, Martín Miguel A
Neuromuscular Unit, Department of Neurology, Hospital Universitario 12 de Octubre, 28041 Madrid, Spain.
Hospital 12 de Octubre Research Institute (imas12), 28041 Madrid, Spain.
J Clin Med. 2021 Dec 22;11(1):22. doi: 10.3390/jcm11010022.
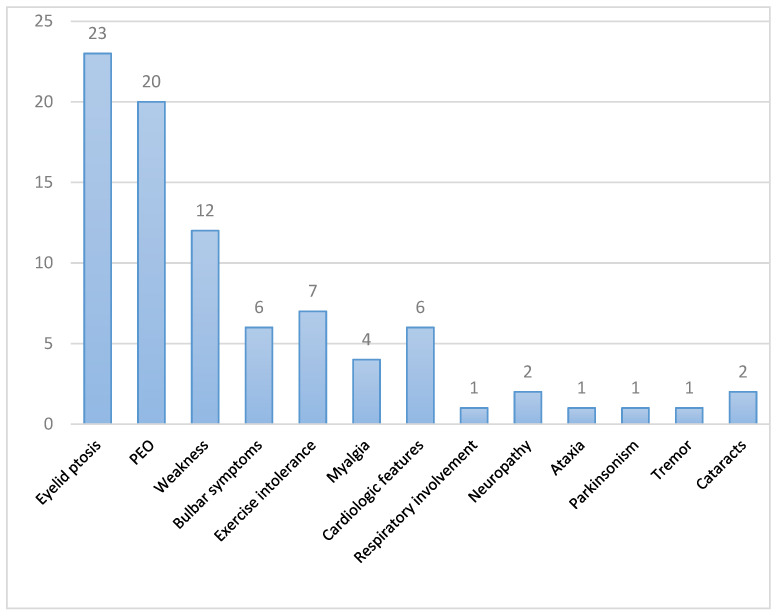
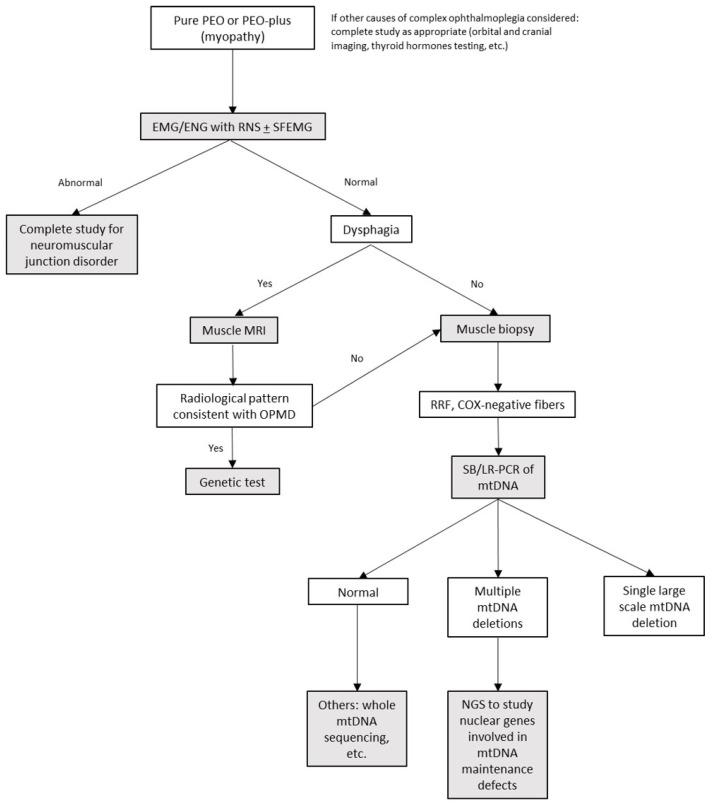
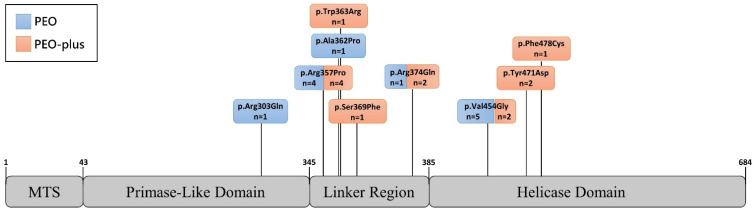
Autosomal dominant mutations in the gene, which encodes a mitochondrial DNA helicase, cause adult-onset progressive external ophthalmoplegia (PEO) and PEO-plus presentations. In this retrospective observational study, we describe clinical and complementary data from 25 PEO patients with mutations in recruited from the Hospital 12 de Octubre Mitochondrial Disorders Laboratory Database. The mean ages of onset and diagnosis were 43 and 63 years, respectively. Family history was positive in 22 patients. Ptosis and PEO (92% and 80%) were the most common findings. Weakness was present in 48%, affecting proximal limbs, neck, and bulbar muscles. Exercise intolerance was present in 28%. Less frequent manifestations were cardiac (24%) and respiratory (4%) involvement, neuropathy (8%), ataxia (4%), and parkinsonism (4%). Only 28% had mild hyperCKemia. All 19 available muscle biopsies showed signs of mitochondrial dysfunction. Ten different mutations were identified, with c.1361T>G (p.Val454Gly) and c.1070G>C (p.Arg357Pro) being the most common. Before definitive genetic confirmation, 56% of patients were misdiagnosed (36% with myasthenia, 20% with oculopharyngeal muscle dystrophy). Accurate differential diagnosis and early confirmation with appropriately chosen complementary studies allow genetic counseling and the avoidance of unnecessary treatments. Thus, mitochondrial myopathies must be considered in PEO/PEO-plus presentations, and particularly, is an important cause when positive family history is present.
该基因编码一种线粒体DNA解旋酶,其常染色体显性突变会导致成人起病的进行性眼外肌麻痹(PEO)及PEO附加型症状。在这项回顾性观察研究中,我们描述了从12月12日医院线粒体疾病实验室数据库招募的25例患有该基因突变的PEO患者的临床及补充数据。发病和诊断的平均年龄分别为43岁和63岁。22例患者有家族史。上睑下垂和PEO(分别为92%和80%)是最常见的表现。48%的患者存在肌无力,累及近端肢体、颈部和延髓肌。28%的患者有运动不耐受。较少见的表现包括心脏受累(24%)、呼吸受累(4%)、神经病变(8%)、共济失调(4%)和帕金森综合征(4%)。只有28%的患者有轻度高肌酸激酶血症。所有19例可获得的肌肉活检均显示出线粒体功能障碍的迹象。共鉴定出10种不同的该基因突变,其中c.1361T>G(p.Val454Gly)和c.1070G>C(p.Arg357Pro)最为常见。在进行明确的基因确诊之前,56%的患者被误诊(36%被误诊为重症肌无力,20%被误诊为眼咽型肌营养不良)。通过适当选择补充检查进行准确的鉴别诊断和早期确诊,可为遗传咨询提供依据,并避免不必要的治疗。因此,在PEO/PEO附加型症状中必须考虑线粒体肌病,特别是当有阳性家族史时,该基因是一个重要病因。