Department of Biochemistry and Molecular Biology, Virginia Commonwealth University, Richmond, VA 23298, USA.
Genzada Pharmaceuticals, Sterling, KS 67579, USA.
Oncotarget. 2022 Feb 4;13:281-290. doi: 10.18632/oncotarget.28189. eCollection 2022.
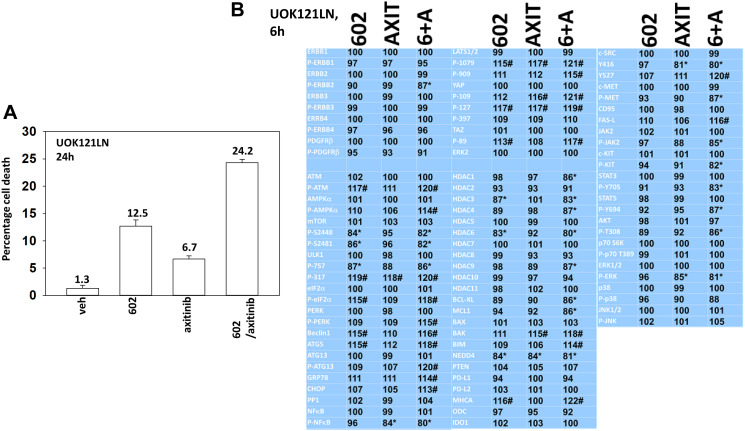
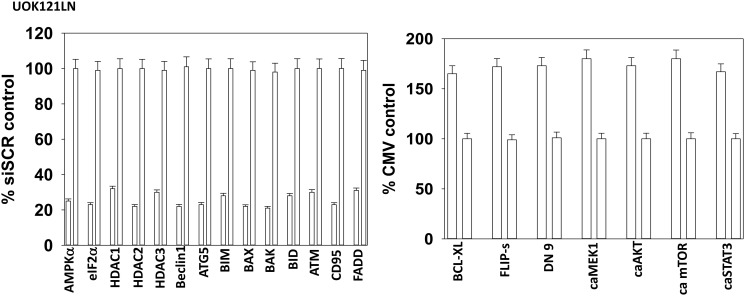
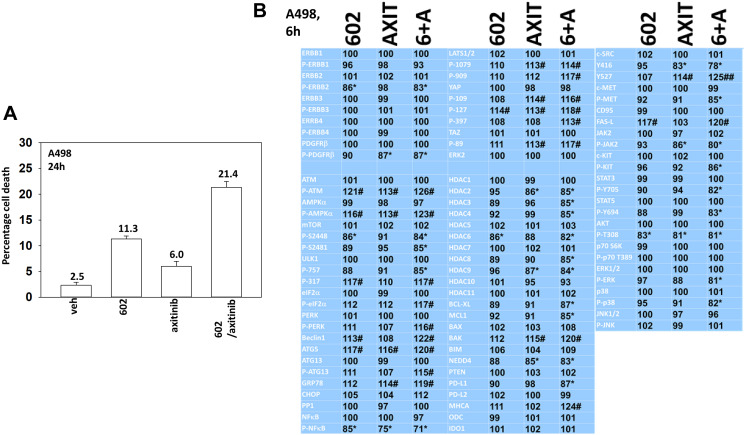
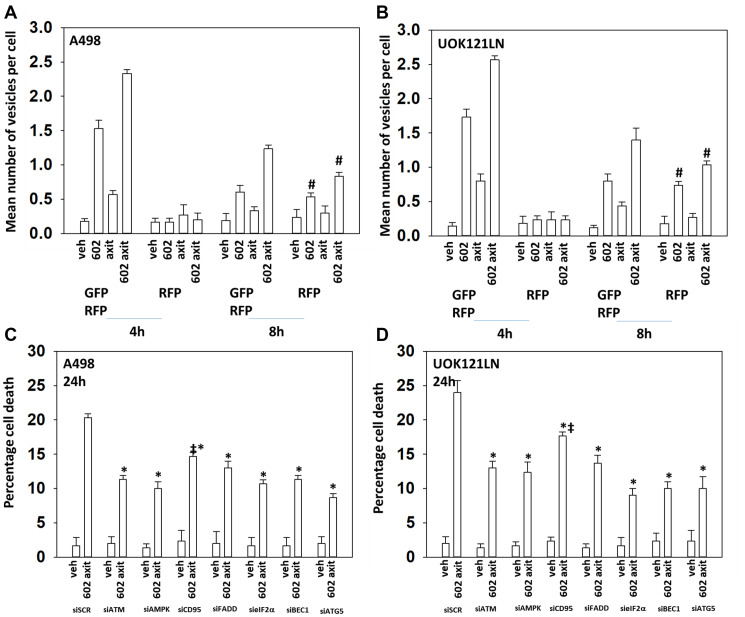
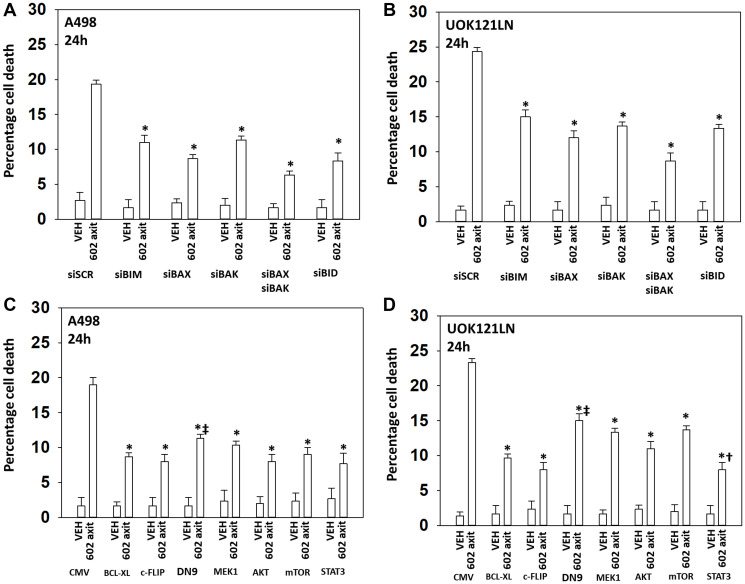
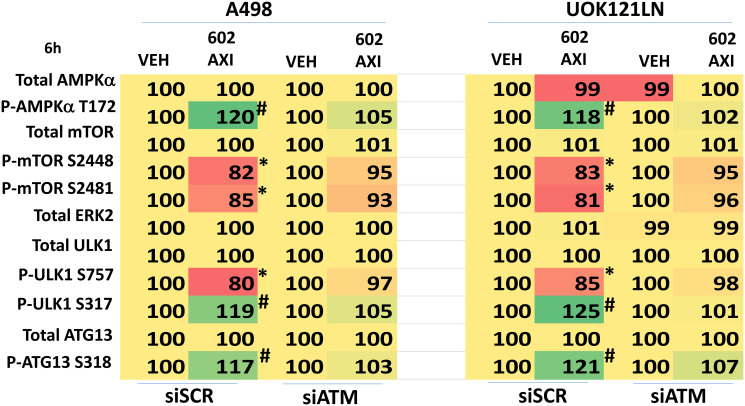
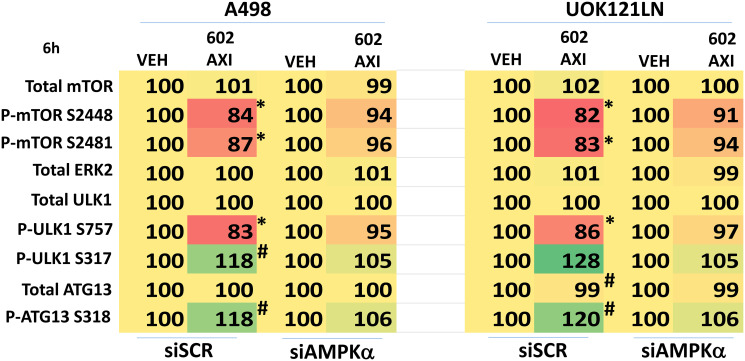
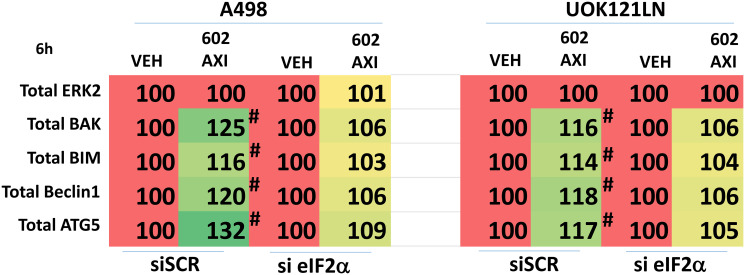
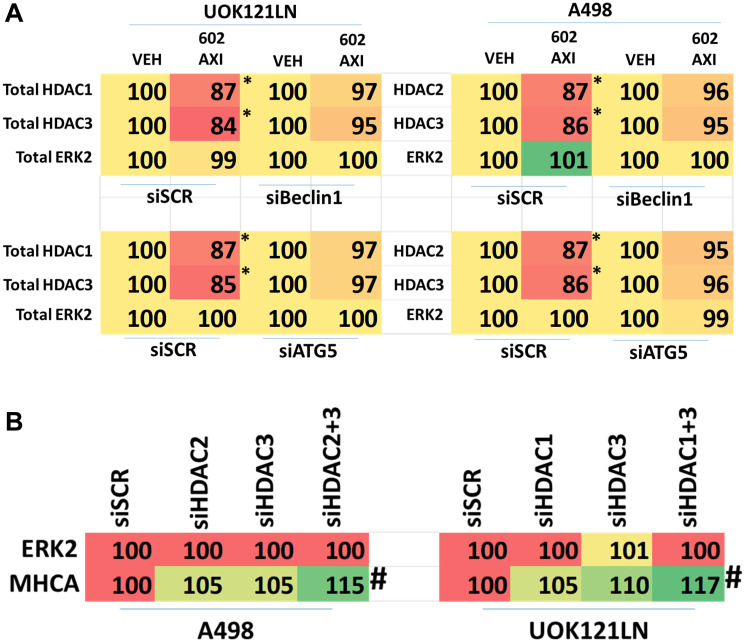
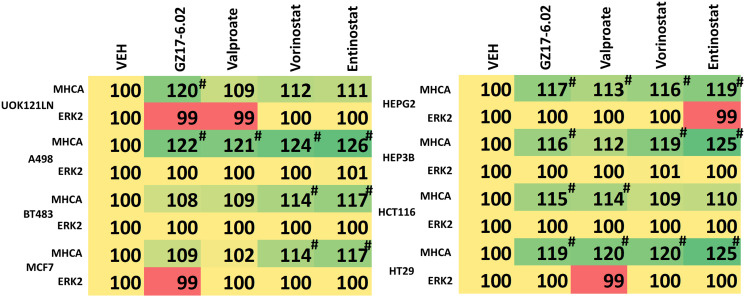
GZ17-6.02 is undergoing clinical evaluation in solid tumors and lymphoma. The present studies were performed to define its biology in renal carcinoma cells and to determine whether it interacted with axitinib to enhance tumor cell killing. GZ17-6.02 interacted in an arithmetically greater than additive fashion with axitinib to kill kidney cancer cells. GZ17-6.02 and axitinib cooperated to inactivate ERBB2, c-MET, c-KIT, c-SRC, the AMPK, STAT3, STAT5 and eIF2α and to activate PERK, ULK1 and ATG13. The drugs interacted to increase the expression of FAS-L and to decrease the levels of MCL1, BCL-XL, and HDACs 1-3. The drugs as single agents inactivated the Hippo pathway. GZ17-6.02 and axitinib interacted to enhance autophagosome formation and autophagic flux. Knock down of Beclin1, ATG5, eIF2α, toxic BH3 domain proteins or CD95/FADD significantly reduced drug combination lethality. GZ17-6.02 and axitinib increased the expression of BAK, BIM, Beclin1 and ATG5, effects blocked by knock down of eIF2α. The drugs increased phosphorylation of ULK1 S757 and ATG13 S318 and decreased the phosphorylation of mTORC1 and mTORC2, effects blocked by knock down of AMPKα. Knock down of Beclin1 or ATG5 prevented the drug combination reducing expression of HDACs 1-3 and from enhancing the expression of MHCA. Knock down of HDACs 1-3 enhanced MHCA expression. We conclude that GZ17-6.02 and axitinib interact to kill requiring ER stress signaling, autophagy and death receptor signaling. Autophagic degradation of HDACs played a key role in enhancing MHCA expression and of a potential improved response to checkpoint inhibitory immunotherapy.
GZ17-6.02 正在进行实体瘤和淋巴瘤的临床评估。本研究旨在确定其在肾癌细胞中的生物学特性,并确定其是否与阿昔替尼相互作用以增强肿瘤细胞杀伤作用。GZ17-6.02 与阿昔替尼以算术方式相互作用,以杀死肾癌细胞。GZ17-6.02 和阿昔替尼合作使 ERBB2、c-MET、c-KIT、c-SRC、AMPK、STAT3、STAT5 和 eIF2α失活,并激活 PERK、ULK1 和 ATG13。这些药物相互作用增加了 FAS-L 的表达,降低了 MCL1、BCL-XL 和 HDACs 1-3 的水平。这些药物作为单一药物使 Hippo 通路失活。GZ17-6.02 和阿昔替尼相互作用增强自噬体形成和自噬流。Beclin1、ATG5、eIF2α、毒性 BH3 结构域蛋白或 CD95/FADD 的敲低显著降低了药物组合的致死性。GZ17-6.02 和阿昔替尼增加了 BAK、BIM、Beclin1 和 ATG5 的表达,这些作用被 eIF2α 的敲低所阻断。这些药物增加了 ULK1 S757 和 ATG13 S318 的磷酸化,降低了 mTORC1 和 mTORC2 的磷酸化,这些作用被 AMPKα 的敲低所阻断。Beclin1 或 ATG5 的敲低阻止了药物组合降低 HDACs 1-3 的表达,并增强了 MHCA 的表达。HDACs 1-3 的敲低增强了 MHCA 的表达。我们得出结论,GZ17-6.02 和阿昔替尼相互作用杀死需要 ER 应激信号、自噬和死亡受体信号的细胞。HDACs 的自噬降解在增强 MHCA 表达和提高对检查点抑制免疫治疗的反应方面发挥了关键作用。