Fang Jia, Tong Zhongyi, Lu Wei
Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, China.
Department of Pathology, Second Xiangya Hospital, Central South University, Changsha, China.
Front Neurol. 2022 Jan 28;13:806224. doi: 10.3389/fneur.2022.806224. eCollection 2022.
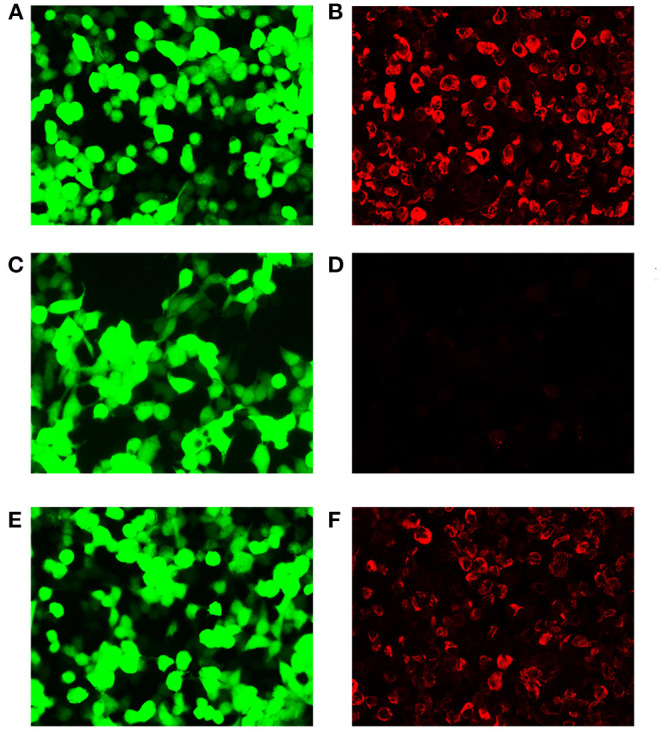
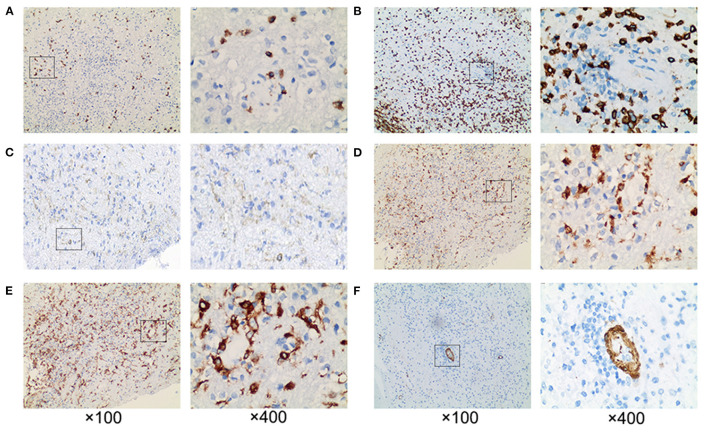
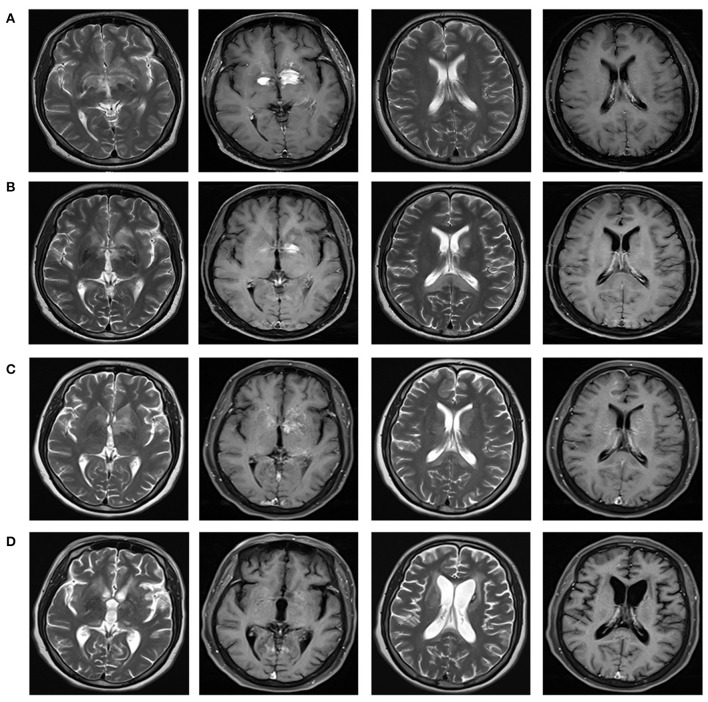
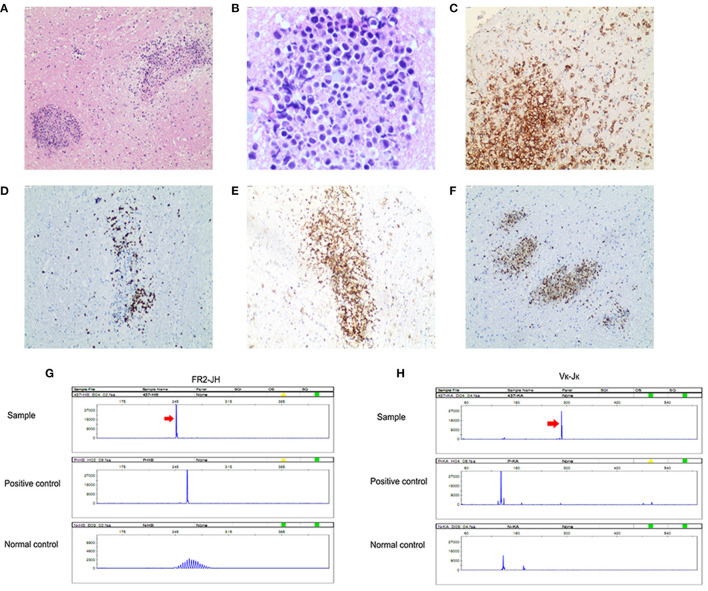
We reported a case of primary central nervous system lymphoma (PCNSL) coexistent with glial fibrillary acidic protein (GFAP) astrocytopathy, and discussed the problems needing attention in the diagnosis and differential diagnosis of GFAP astrocytopathy. Our patient was a 51-year-old female who presented with somnolence for a month, and memory declination for 10 days. Brain magnetic resonance imaging (MRI) demonstrated multiple abnormal enhancement lesions in bilateral basal ganglia and around the third ventricle, as well as transient T2-weighted hyper-intensity lesions at the splenium of the corpus callosum during the course of the disease. The cerebrospinal fluid (CSF) was positive for anti-GFAP antibodies by antigen-transfected HEK293 cell-based assay (indirect immunofluorescence assay). She was initially diagnosed with autoimmune GFAP astrocytopathy. After treatment with corticosteroids for about 2 months, she displayed poor response and even worsened clinical manifestations when the dose of prednisone reduced to 45 mg. Stereotactic brain biopsy was adopted and the diagnosis of large B-cell lymphoma, non-germinal center type was established on pathological examination. The results of brain biopsy also showed perivascular inflammation and CD8+ T cell infiltration, which also accorded with GFAP astrocytopathy. After chemotherapy with rituximab and methotrexate, the patient showed clinical and radiological improvement significantly. Our findings suggest that positivity of GFAP antibody calls for cautious interpretation. Cancer screening appropriate for age, sex, and risk factors is recommended for GFAP antibody-positive patients, especially for patients with atypical clinical and radiologic manifestations.
我们报告了一例原发性中枢神经系统淋巴瘤(PCNSL)合并胶质纤维酸性蛋白(GFAP)星形细胞病的病例,并讨论了GFAP星形细胞病诊断和鉴别诊断中需要注意的问题。我们的患者是一名51岁女性,出现嗜睡1个月,记忆力减退10天。脑磁共振成像(MRI)显示双侧基底节及第三脑室周围有多个异常强化病灶,病程中胼胝体压部有短暂的T2加权高信号病灶。通过基于抗原转染的HEK293细胞的检测(间接免疫荧光检测),脑脊液(CSF)抗GFAP抗体呈阳性。她最初被诊断为自身免疫性GFAP星形细胞病。用皮质类固醇治疗约2个月后,她反应不佳,当泼尼松剂量减至45 mg时,临床表现甚至恶化。采用立体定向脑活检,病理检查确诊为大B细胞淋巴瘤,非生发中心型。脑活检结果还显示血管周围炎症和CD8 + T细胞浸润,这也符合GFAP星形细胞病。用利妥昔单抗和甲氨蝶呤化疗后,患者临床和影像学表现明显改善。我们的研究结果表明,GFAP抗体阳性需要谨慎解读。对于GFAP抗体阳性患者,尤其是临床表现和影像学表现不典型的患者,建议根据年龄、性别和危险因素进行适当的癌症筛查。