Zhuang Dan-Yan, Ding Shu-Xia, Wang Fei, Yang Xiang-Chun, Pan Xiao-Li, Bao You-Wei, Zhou Li-Ming, Li Hai-Bo
The Central Laboratory of Birth Defects Prevention and Control, Ningbo Women and Children's Hospital, Ningbo, China.
Department of Endocrinology and Genetic Metabolism of Pediatrics, Ningbo Women and Children's Hospital, Ningbo, China.
Front Genet. 2022 Jan 28;12:791869. doi: 10.3389/fgene.2021.791869. eCollection 2021.
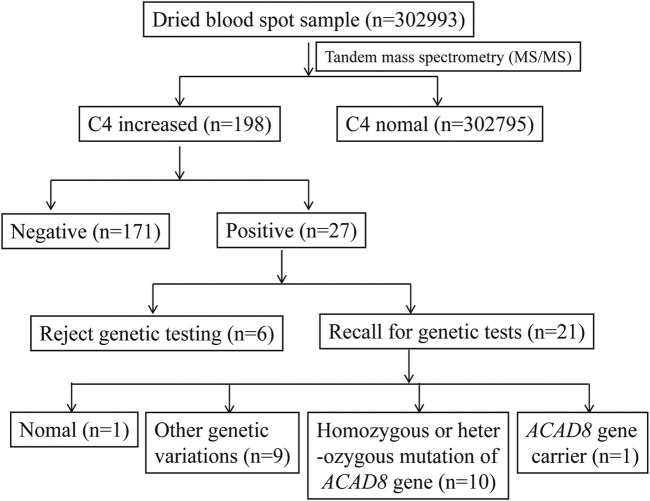
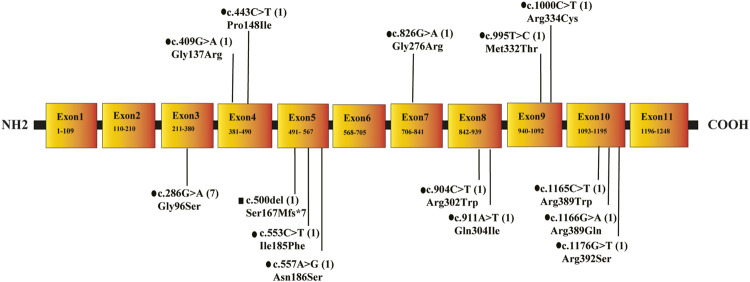
Isobutyryl-CoA dehydrogenase deficiency (IBDHD, MIM: #611283) is a rare autosomal recessive hereditary disease, which is caused by genetic mutations of acyl-CoA dehydrogenase (ACAD) 8 and associated with valine catabolism. Here, tandem mass spectrometry (MS/MS) was applied to screen 302,993 neonates for inherited metabolic diseases (IMD) in Ningbo of China from 2017 to 2020. The results suggest that 198 newborns (0.7‰) were initially screened positive for IBDHD with C4-Carnitine, and 27 cases (0.1‰) were re-screened positive. Genetic diagnosis was performed on 21 of the 27 cases. Seven compound heterozygous variations, three biallelic variations, and one heterozygous variation of ACAD8 were found with a pathogenicity rate of 33.3% (7/21). In addition, seven biallelic variations, one heterozygous variation of acyl-CoA dehydrogenase short chain (), and one biallelic variation of acyl-CoA dehydrogenase short/branched chain () was detected. Further research showed that mutations of 11 IBDHD cases distributed in six different exons with total 14 mutation sites. Five of which were known suspected pathogenic sites (c.286G > A, c.553C > T, c.1000C > T, c.409G > A, c.500del) and six were novel mutation sites: c.911A > T, c.904C > T, c.826G > A, c.995T > C, c.1166G > A, c.1165C > T. This finding enriched the mutation spectrum of in IBDHD.
异丁酰辅酶A脱氢酶缺乏症(IBDHD,MIM:#611283)是一种罕见的常染色体隐性遗传性疾病,由酰基辅酶A脱氢酶(ACAD)8基因突变引起,与缬氨酸分解代谢有关。在此,应用串联质谱(MS/MS)对2017年至2020年中国宁波的302,993名新生儿进行遗传性代谢疾病(IMD)筛查。结果显示,198名新生儿(0.7‰)初筛C4-肉碱IBDHD呈阳性,27例(0.1‰)复筛呈阳性。对27例中的21例进行了基因诊断。发现7种复合杂合变异、3种双等位基因变异和1种ACAD8杂合变异,致病率为33.3%(7/21)。此外,检测到7种双等位基因变异、1种短链酰基辅酶A脱氢酶()杂合变异和1种短/支链酰基辅酶A脱氢酶()双等位基因变异。进一步研究表明,11例IBDHD病例的突变分布在6个不同外显子中,共有14个突变位点。其中5个是已知的疑似致病位点(c.286G > A、c.553C > T、c.1000C > T、c.409G > A、c.500del),6个是新的突变位点:c.911A > T、c.904C > T、c.826G > A、c.995T > C、c.1166G > A、c.1165C > T。这一发现丰富了IBDHD中ACAD8的突变谱。