Neuroscience and Mental Health Institute, Faculty of Medicine and Dentistry, University of Alberta, 116 St. and 85 Ave., Edmonton, AB T6G 2E1, Canada.
Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, 116 St. and 85 Ave., Edmonton, AB T6G 2E1, Canada.
Cells. 2022 Jan 26;11(3):417. doi: 10.3390/cells11030417.
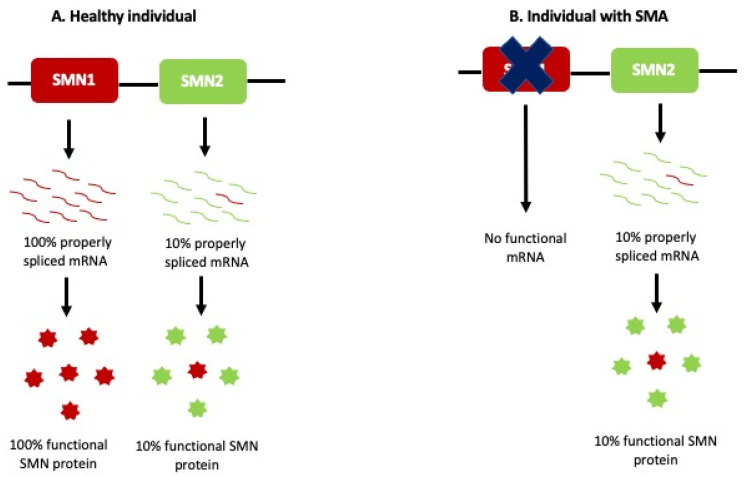
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder and one of the most common genetic causes of infant death. It is characterized by progressive weakness of the muscles, loss of ambulation, and death from respiratory complications. SMA is caused by the homozygous deletion or mutations in the survival of the motor neuron 1 () gene. Humans, however, have a nearly identical copy of known as the gene. The severity of the disease correlates inversely with the number of copies present. cannot completely compensate for the loss of in SMA patients because it can produce only a fraction of functional SMN protein. SMN protein is ubiquitously expressed in the body and has a variety of roles ranging from assembling the spliceosomal machinery, autophagy, RNA metabolism, signal transduction, cellular homeostasis, DNA repair, and recombination. Motor neurons in the anterior horn of the spinal cord are extremely susceptible to the loss of SMN protein, with the reason still being unclear. Due to the ability of the gene to produce small amounts of functional SMN, two FDA-approved treatment strategies, including an antisense oligonucleotide (AON) nusinersen and small-molecule risdiplam, target to produce more functional SMN. On the other hand, Onasemnogene abeparvovec (brand name Zolgensma) is an FDA-approved adeno-associated vector 9-mediated gene replacement therapy that can deliver a copy of the human In this review, we summarize the SMA etiology, the role of SMN, and discuss the challenges of the therapies that are approved for SMA treatment.
脊髓性肌萎缩症(SMA)是一种常染色体隐性神经退行性疾病,也是婴儿死亡的最常见遗传原因之一。它的特征是肌肉逐渐无力、丧失行动能力,并因呼吸并发症而死亡。SMA 是由运动神经元生存基因 1(SMN1)的纯合缺失或突变引起的。然而,人类有一个几乎相同的 SMN1 副本,称为 SMN2 基因。疾病的严重程度与存在的 SMN2 拷贝数呈反比相关。SMN2 不能完全弥补 SMA 患者中 SMN1 的缺失,因为它只能产生功能性 SMN 蛋白的一小部分。SMN 蛋白在体内广泛表达,具有多种作用,包括组装剪接体机制、自噬、RNA 代谢、信号转导、细胞内稳态、DNA 修复和重组。脊髓前角的运动神经元对 SMN 蛋白的缺失极其敏感,其原因尚不清楚。由于 SMN2 基因能够产生少量功能性 SMN,两种已获 FDA 批准的治疗策略,包括反义寡核苷酸(AON)nusinersen 和小分子 risdiplam,都针对 SMN2 以产生更多功能性 SMN。另一方面,onasemnogene abeparvovec(商品名 Zolgensma)是一种已获 FDA 批准的腺相关病毒 9 介导的基因替代疗法,可递送人类 SMN1 的一个拷贝。在这篇综述中,我们总结了 SMA 的病因、SMN 的作用,并讨论了已批准用于 SMA 治疗的疗法所面临的挑战。