Li Tianlong, Sun Haibin, Li Yiming, Su Lianjiu, Jiang Jun, Liu Ye, Jiang Nanhui, Huang Rong, Zhang Jiahao, Peng Zhiyong
Department of Critical Care Medicine, Zhongnan Hospital of Wuhan University, Wuhan, Hubei province, 430071, China.
Center of Critical Nephrology, Department of Critical Care Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA, 15223, USA.
Cell Death Discov. 2022 Feb 14;8(1):61. doi: 10.1038/s41420-022-00859-z.
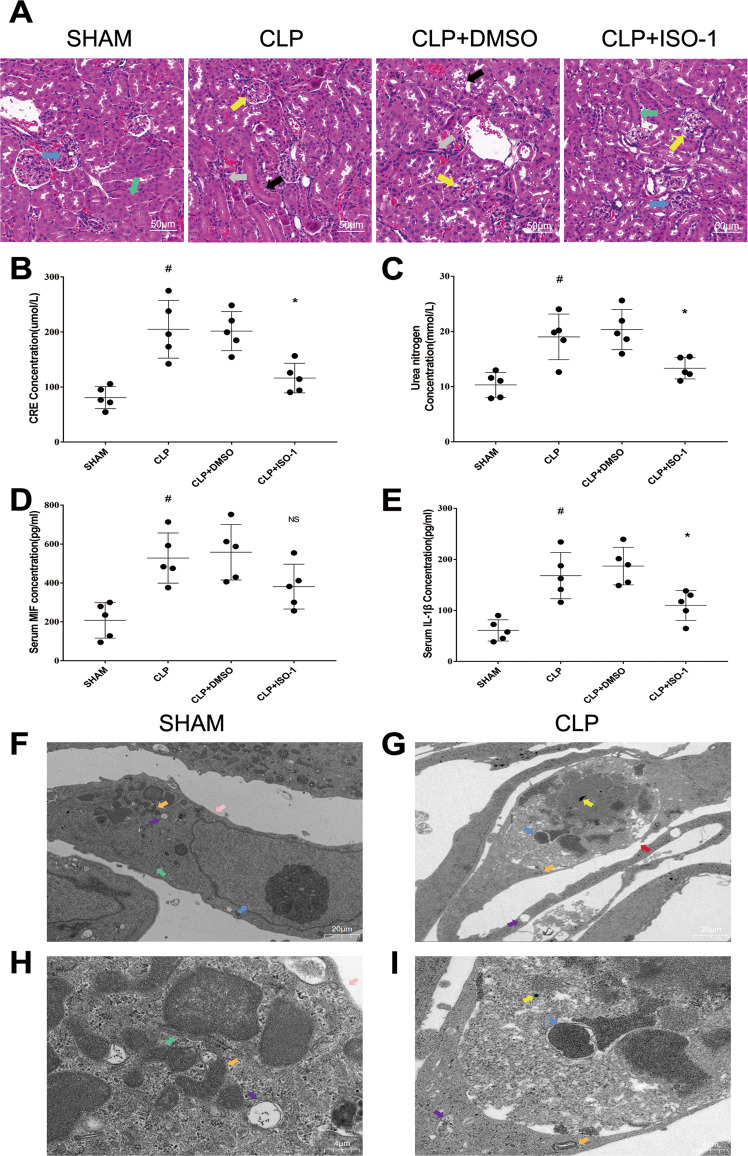
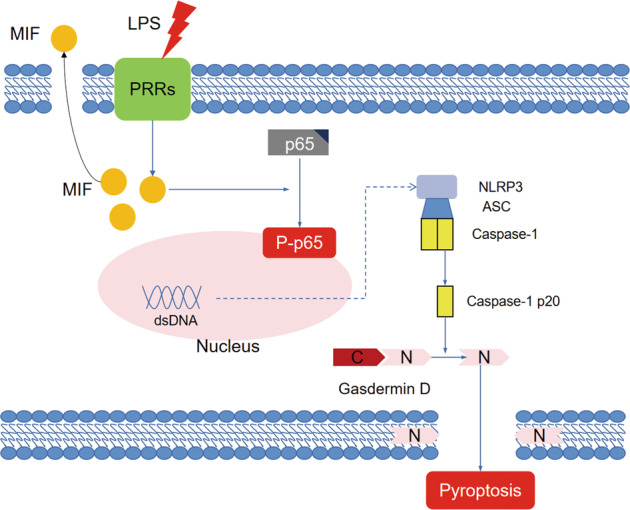
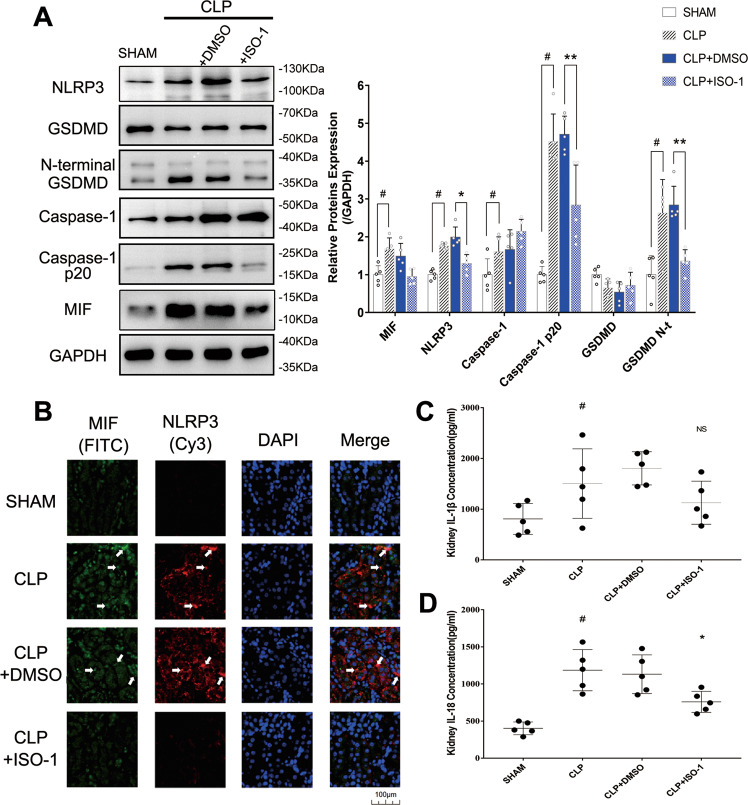
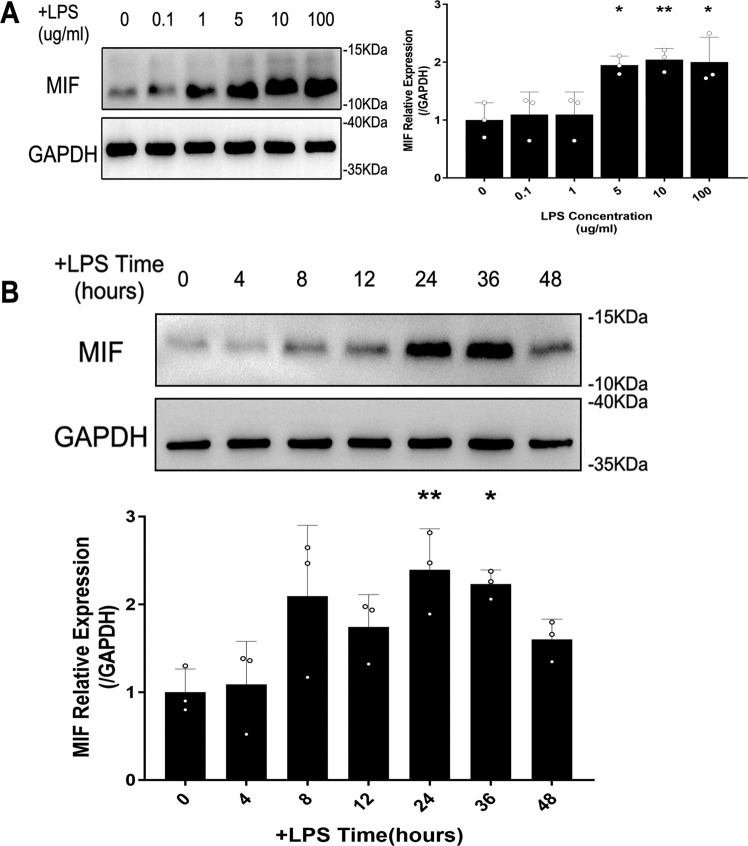
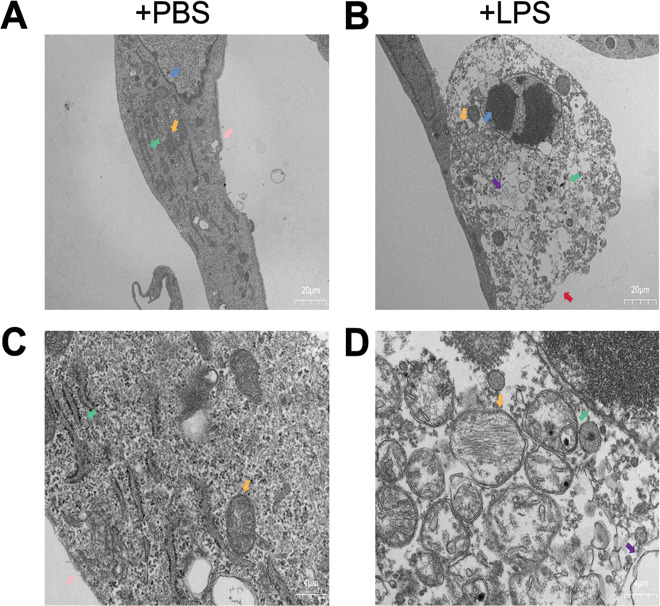
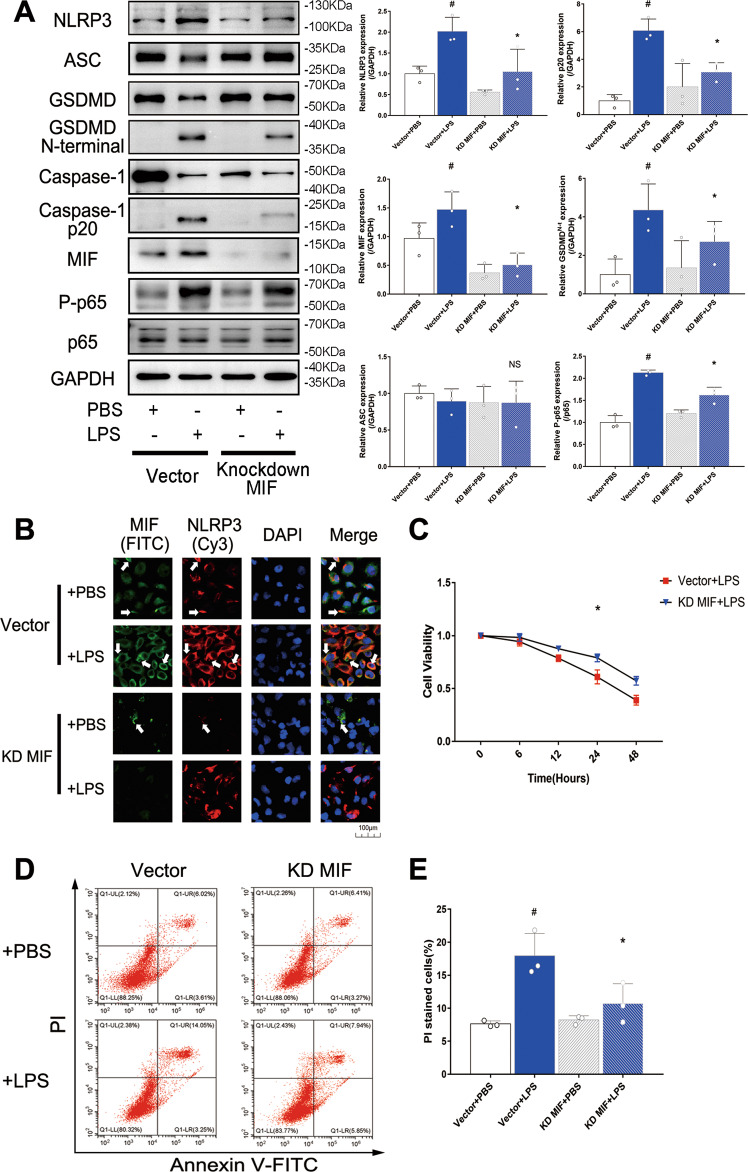
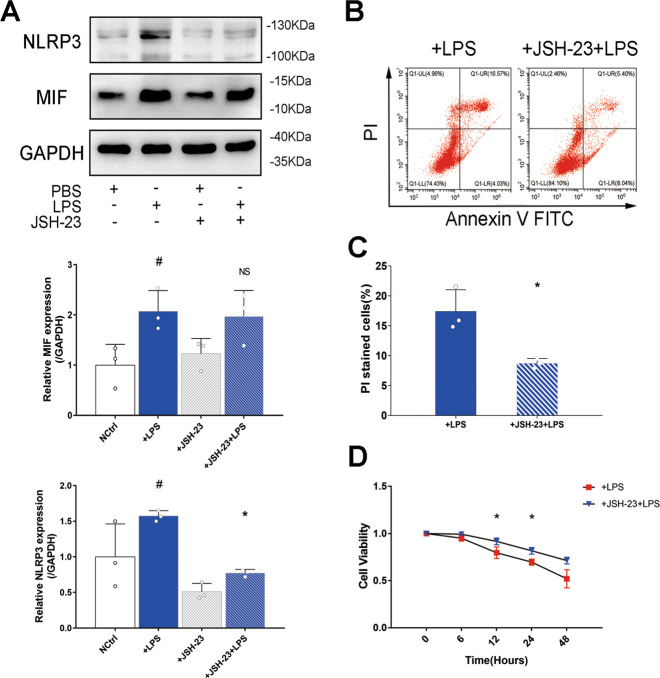
Sepsis-induced AKI (acute kidney injury) is considered an inflammation-related disease with high mortality. LPS-induced (Lipopolysaccharide) TLR4-NFκB pathway activation plays an important role in sepsis-induced AKI. Pyroptosis closely associated with inflammation response includes inflammasome formation, caspase1 activation and GSDMD N-terminal fragment cleavage that leads to cell membrane rupture and cell death, which may be related to the pathogenesis of sepsis-induced AKI. MIF (Macrophage migration inhibitory factor), associated with inflammation response, has been proved as a biomarker of sepsis, and perhaps regulate pyroptosis in sepsis-induced AKI. In this study, we focus on investigating the mechanism of MIF promoting pyroptosis in sepsis-induced AKI. MIF and pyroptosis-related proteins were up-regulated in kidney tissue of mice with CLP (cecum ligation puncture) surgery and in LPS-injured human kidney-2 (HK-2) cells. NLRP3 was down-regulated following the suppression of MIF topoisomerase activity by ISO-1 in kidney tissue of CLP mice. Knockdown of MIF alleviated NLRP3 inflammasome mediated pyroptosis in LPS-injured HK-2 cells. Meanwhile, we noted that phosphorylation of p65 was down-regulated by knockdown of MIF. Up-regulation of NLRP3 in response to LPS stimulation could be reversed by JSH-23, an inhibitor of NFκB pathway, but MIF was not affected. In conclusion, up-regulation of MIF in sepsis-induced AKI shows a renal damaged effect that aggravates NLRP3 inflammasome mediated cell pyroptosis through promoting phosphorylation of p65. This study demonstrated a novel mechanism of MIF regulating NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI.
脓毒症诱导的急性肾损伤(AKI)被认为是一种死亡率很高的炎症相关疾病。脂多糖(LPS)诱导的Toll样受体4(TLR4)-核因子κB(NFκB)信号通路激活在脓毒症诱导的AKI中起重要作用。与炎症反应密切相关的细胞焦亡包括炎性小体形成、半胱天冬酶1激活和Gasdermin D(GSDMD)N端片段切割,导致细胞膜破裂和细胞死亡,这可能与脓毒症诱导的AKI发病机制有关。巨噬细胞移动抑制因子(MIF)与炎症反应相关,已被证明是脓毒症的生物标志物,并且可能在脓毒症诱导的AKI中调节细胞焦亡。在本研究中,我们着重探讨MIF促进脓毒症诱导的AKI中细胞焦亡的机制。在盲肠结扎穿孔(CLP)手术小鼠的肾组织以及LPS损伤的人肾2(HK-2)细胞中,MIF和细胞焦亡相关蛋白表达上调。在CLP小鼠肾组织中,异戊巴比妥(ISO-1)抑制MIF拓扑异构酶活性后,NLRP3表达下调。敲低MIF可减轻LPS损伤的HK-2细胞中NLRP3炎性小体介导的细胞焦亡。同时,我们注意到敲低MIF可使p65磷酸化水平下调。NFκB信号通路抑制剂JSH-23可逆转LPS刺激引起的NLRP3上调,但对MIF无影响。总之,脓毒症诱导的AKI中MIF上调具有肾损伤作用,通过促进p65磷酸化加重NLRP3炎性小体介导的细胞焦亡。本研究揭示了MIF在脓毒症诱导的AKI中调节NLRP3炎性小体介导的细胞焦亡的新机制。