Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA.
Department of Genetics, Evolution and Environment, University College London, London WC1E 6BT, UK.
Syst Biol. 2022 Aug 10;71(5):1159-1177. doi: 10.1093/sysbio/syac009.
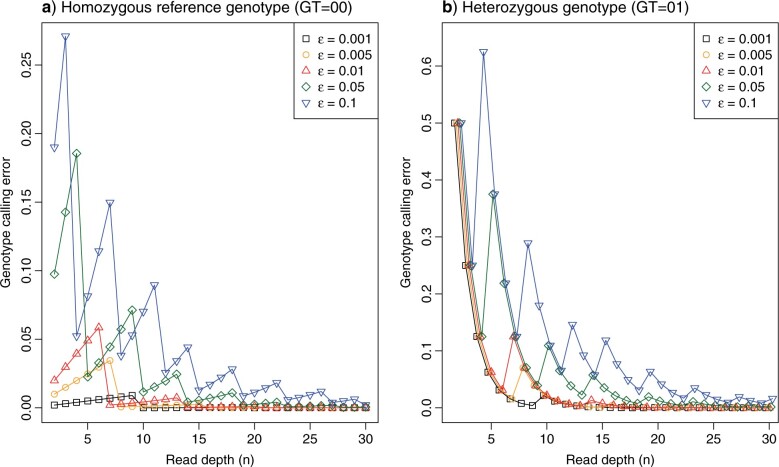
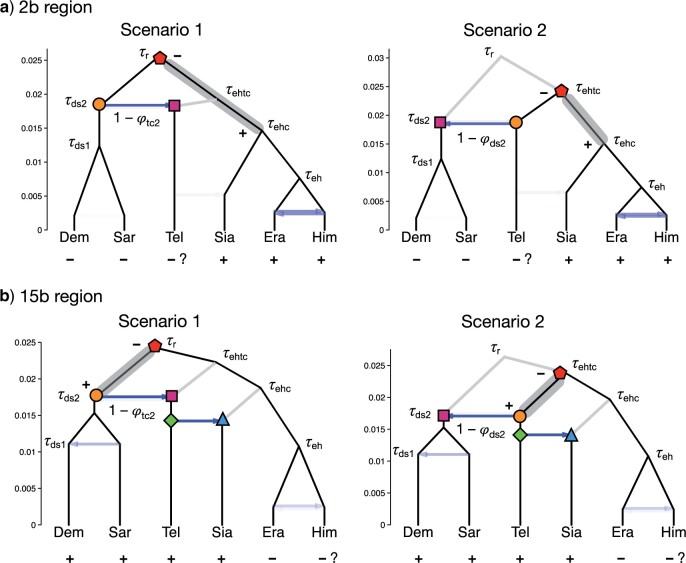
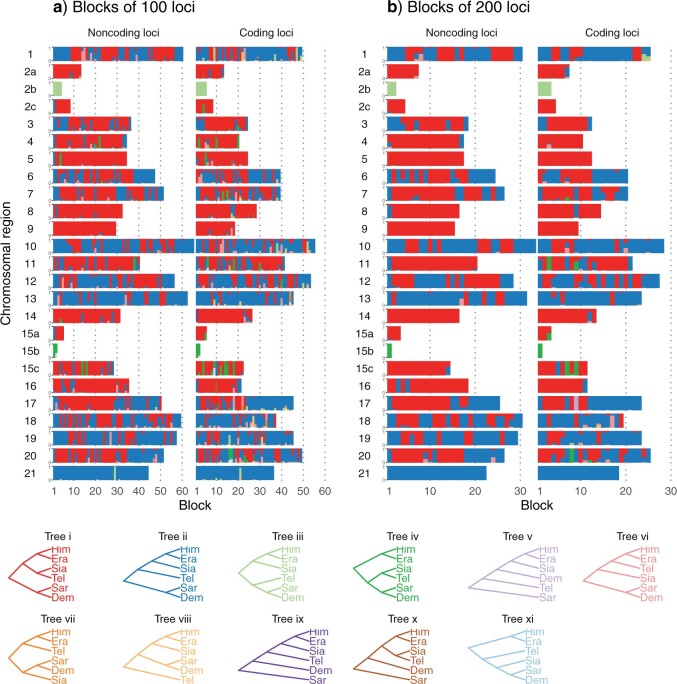
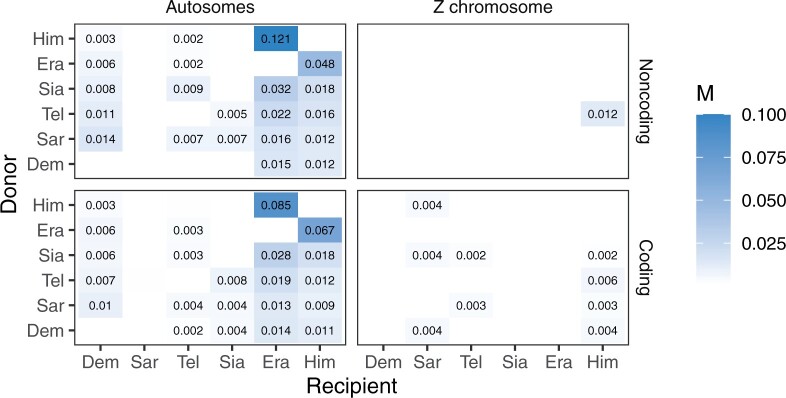
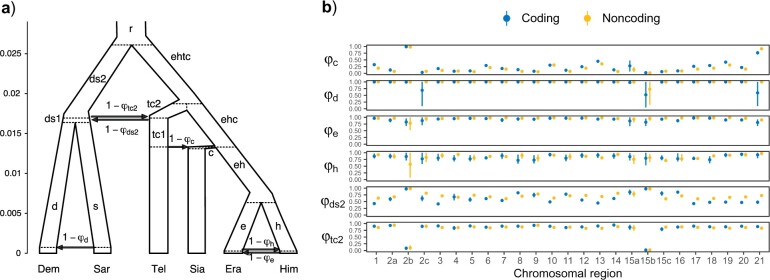
Introgressive hybridization plays a key role in adaptive evolution and species diversification in many groups of species. However, frequent hybridization and gene flow between species make estimation of the species phylogeny and key population parameters challenging. Here, we show that by accounting for phasing and using full-likelihood methods, introgression histories and population parameters can be estimated reliably from whole-genome sequence data. We employ the multispecies coalescent (MSC) model with and without gene flow to infer the species phylogeny and cross-species introgression events using genomic data from six members of the erato-sara clade of Heliconius butterflies. The methods naturally accommodate random fluctuations in genealogical history across the genome due to deep coalescence. To avoid heterozygote phasing errors in haploid sequences commonly produced by genome assembly methods, we process and compile unphased diploid sequence alignments and use analytical methods to average over uncertainties in heterozygote phase resolution. There is robust evidence for introgression across the genome, both among distantly related species deep in the phylogeny and between sister species in shallow parts of the tree. We obtain chromosome-specific estimates of key population parameters such as introgression directions, times and probabilities, as well as species divergence times and population sizes for modern and ancestral species. We confirm ancestral gene flow between the sara clade and an ancestral population of Heliconius telesiphe, a likely hybrid speciation origin for Heliconius hecalesia, and gene flow between the sister species Heliconius erato and Heliconius himera. Inferred introgression among ancestral species also explains the history of two chromosomal inversions deep in the phylogeny of the group. This study illustrates how a full-likelihood approach based on the MSC makes it possible to extract rich historical information of species divergence and gene flow from genomic data. [3s; bpp; gene flow; Heliconius; hybrid speciation; introgression; inversion; multispecies coalescent].
渐渗杂交在许多物种群体的适应性进化和物种多样化中起着关键作用。然而,物种之间频繁的杂交和基因流动使得估计物种系统发育和关键种群参数具有挑战性。在这里,我们表明,通过考虑相位并使用全似然方法,可以从全基因组序列数据中可靠地估计渐渗历史和种群参数。我们使用带有和不带有基因流动的多物种合并(MSC)模型,使用来自 6 种 Heliconius 蝴蝶的 erato-sara 支系的基因组数据推断物种系统发育和跨物种渐渗事件。该方法自然适应了由于深合并而导致基因组中谱系历史随机波动的情况。为了避免由于基因组组装方法通常产生的单倍体序列中的杂合子相位错误,我们处理和编译未相位的二倍体序列比对,并使用分析方法平均异质相位分辨率的不确定性。有强有力的证据表明整个基因组中存在渐渗现象,包括在系统发育深处的远缘物种之间以及在树浅部的姐妹物种之间。我们获得了关键种群参数的染色体特异性估计,例如渐渗方向、时间和概率,以及现代和祖先物种的物种分歧时间和种群大小。我们证实了 sara 支系与 Heliconius telesiphe 的祖先种群之间的祖先基因流,这可能是 Heliconius hecalesia 杂种形成的起源,以及姐妹物种 Heliconius erato 和 Heliconius himera 之间的基因流。推断出的祖先物种之间的渐渗现象也解释了该群体系统发育中两个染色体倒位的历史。本研究说明了基于 MSC 的全似然方法如何从基因组数据中提取物种分歧和基因流动的丰富历史信息。[3s; bpp; 基因流动; Heliconius; 杂种形成; 渐渗; 倒位; 多物种合并]。