Laboratory of Fish Molecular Immunology, College of Fisheries and Life Science, Shanghai Ocean University, Shanghai, China.
Laboratory of Fish Molecular Immunology, College of Fisheries and Life Science, Shanghai Ocean University, Shanghai, China; Laboratory of Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China; Key Laboratory of Exploration and Utilization of Aquatic Genetic Resources (Shanghai Ocean University), Ministry of Education, Shanghai, China; National Pathogen Collection Center for Aquatic Animals, Shanghai Ocean University, Shanghai, China.
J Biol Chem. 2022 Mar;298(3):101730. doi: 10.1016/j.jbc.2022.101730. Epub 2022 Feb 15.
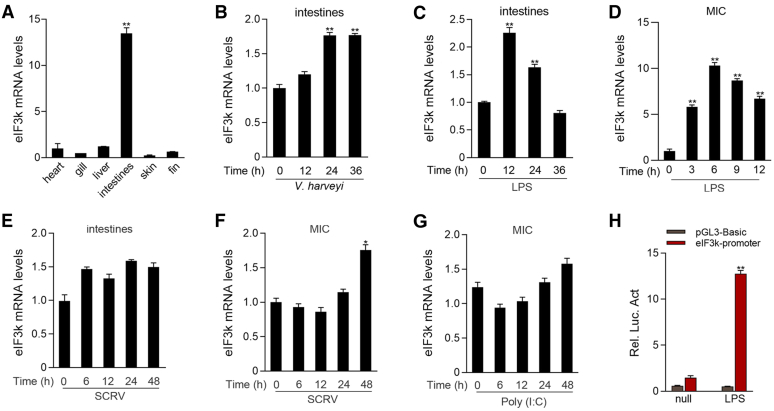
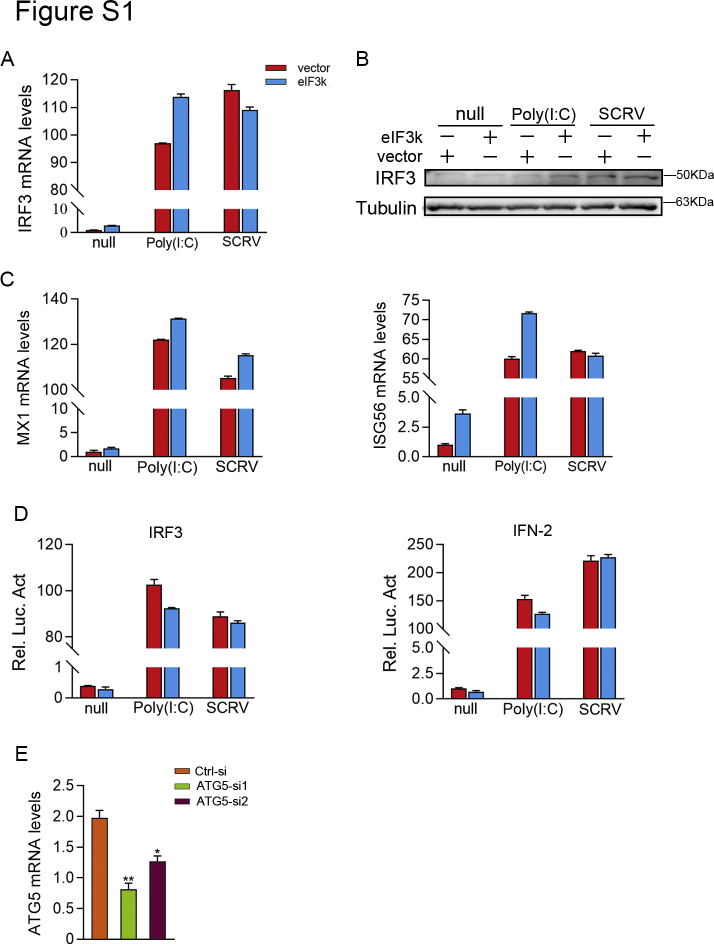
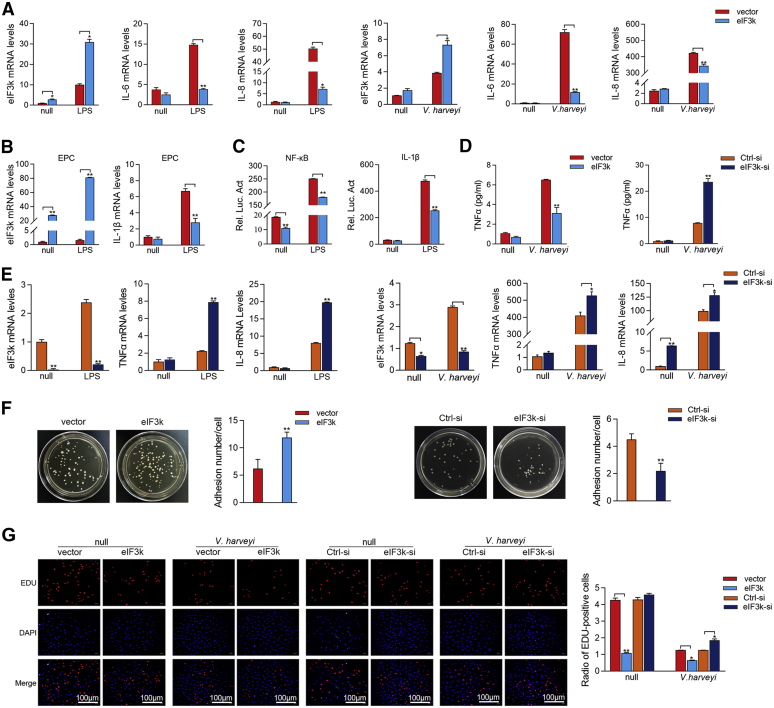
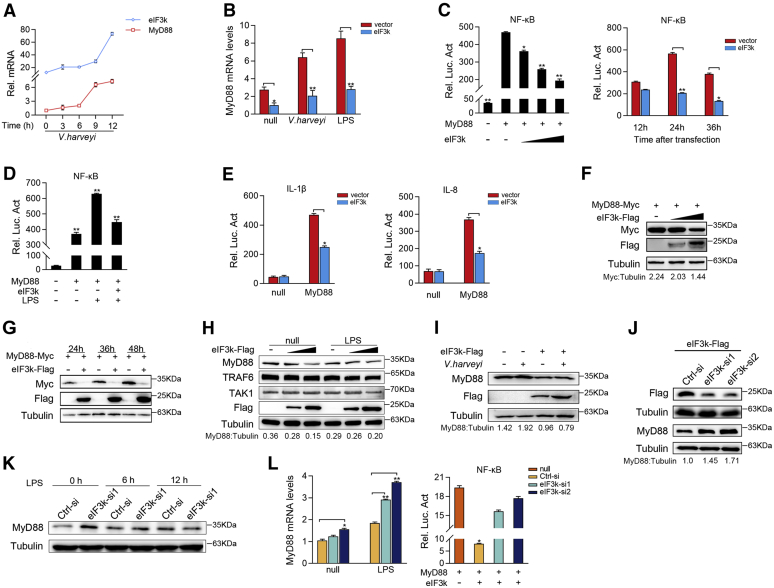
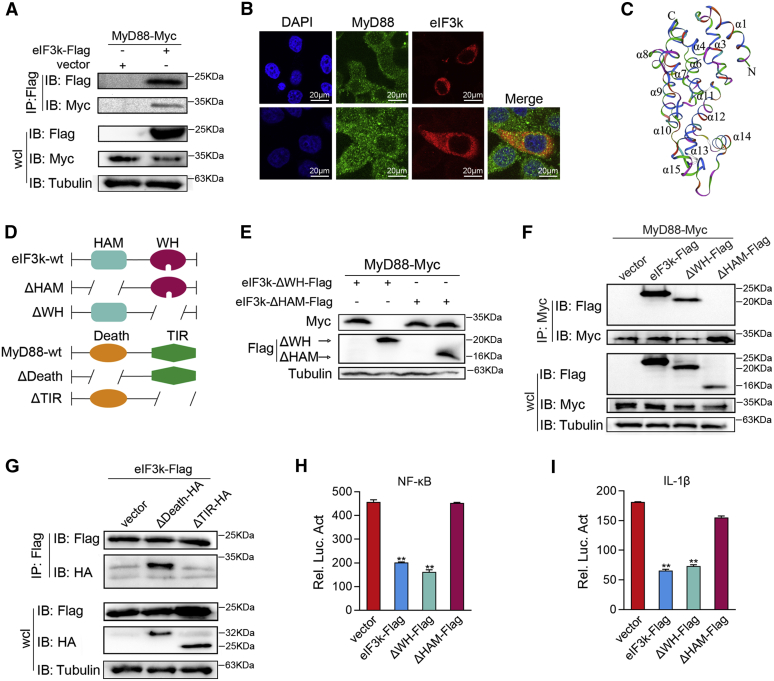
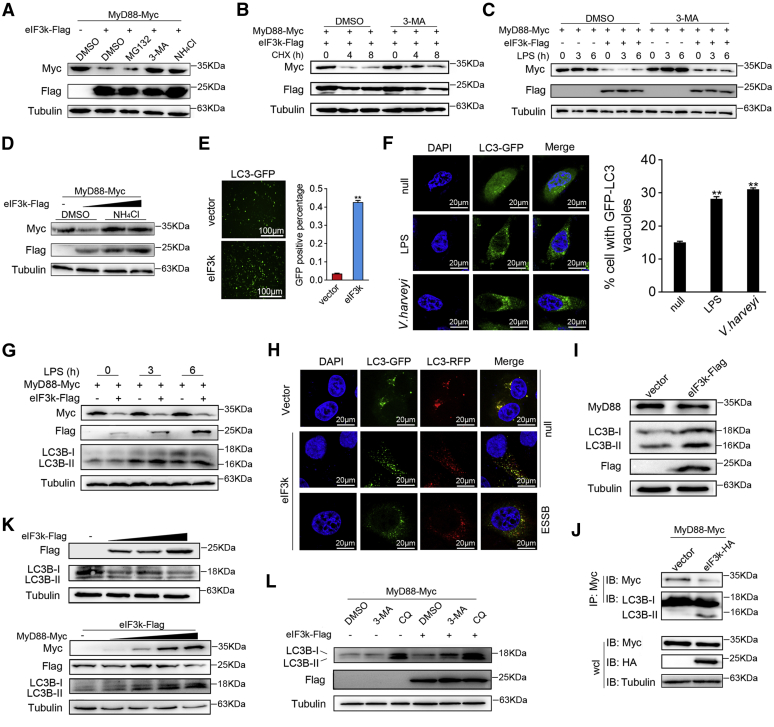
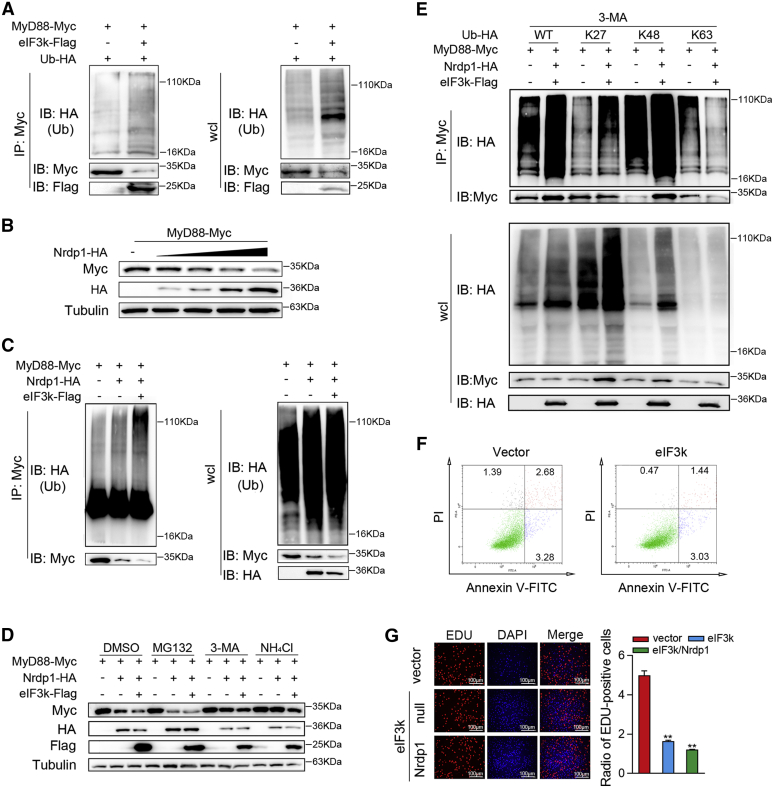
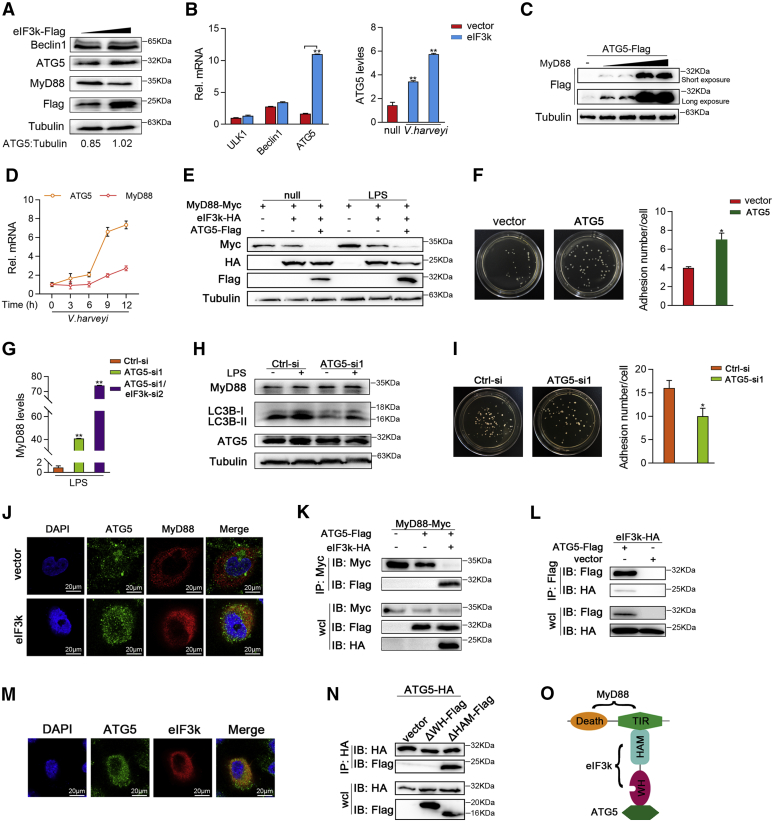
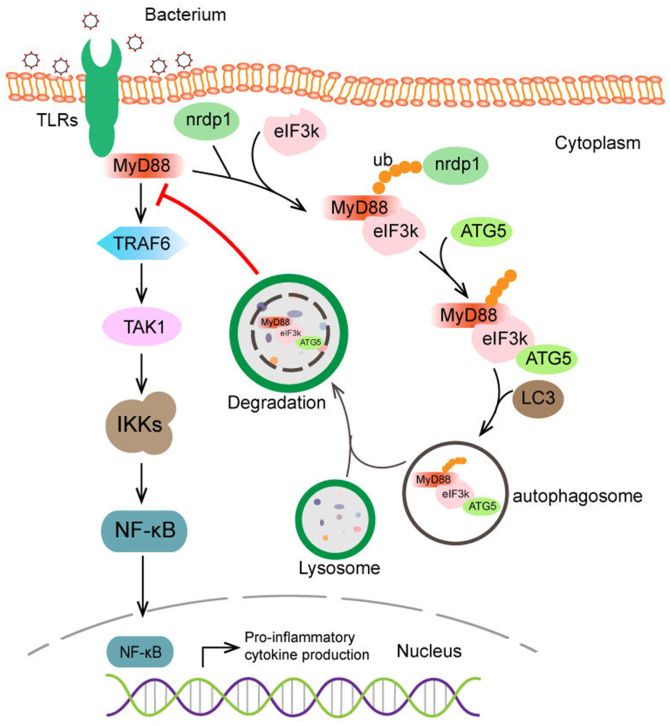
Optimal activation of NF-κB signaling is crucial for the initiation of inflammatory responses and eliminating invading bacteria. Bacteria have likewise evolved the ability to evade immunity; however, mechanisms by which bacteria dysregulate host NF-κB signaling are unclear. In this study, we identify eukaryotic translation initiation factor eIF3k, a nonessential member of the eIF3 translation initiation complex, as a suppressor of the NF-κB pathway. Mechanistically, we show that eIF3k expression induced by Vibrio harveyi enhances E3 ligase Nrdp1-mediated K27-linked ubiquitination of MyD88, an upstream regulator of NF-κB pathway activation. Furthermore, we show that eIF3k acts as a bridge linking ubiquitin-tagged MyD88 and ATG5, an important mediator of autophagy. We demonstrate that the MyD88-eIF3k-ATG5 complex is transported to the autophagosome for degradation, and that innate immune signaling is subsequently terminated and does not attack invading V. harveyi. Therefore, our study identifies eIF3k as a specific inhibitor of the MyD88-dependent NF-κB pathway and suggests that eIF3k may act as a selective autophagic receptor that synergizes with ATG5 to promote the autophagic degradation of MyD88, which helps V. harveyi to evade innate immunity. We conclude that V. harveyi can manipulate a host's autophagy process to evade immunity in fish and also provide a new perspective on mammalian resistance to bacterial invasion.
NF-κB 信号的最佳激活对于炎症反应的启动和消除入侵细菌至关重要。细菌同样进化出了逃避免疫的能力;然而,细菌如何失调宿主 NF-κB 信号的机制尚不清楚。在这项研究中,我们确定了真核翻译起始因子 eIF3k,它是 eIF3 翻译起始复合物的非必需成员,是 NF-κB 途径的抑制剂。从机制上讲,我们表明哈维弧菌诱导的 eIF3k 表达增强了 E3 连接酶 Nrdp1 介导的 NF-κB 途径激活上游调节剂 MyD88 的 K27 连接泛素化。此外,我们表明 eIF3k 作为一个桥接,将泛素化的 MyD88 和自噬的重要介质 ATG5 连接起来。我们证明 MyD88-eIF3k-ATG5 复合物被转运到自噬体进行降解,随后先天免疫信号被终止,不会攻击入侵的 V. harveyi。因此,我们的研究确定 eIF3k 是 MyD88 依赖性 NF-κB 途径的特异性抑制剂,并表明 eIF3k 可能作为一种选择性自噬受体,与 ATG5 协同作用促进 MyD88 的自噬降解,从而帮助 V. harveyi 逃避先天免疫。我们得出结论,V. harveyi 可以操纵宿主的自噬过程来逃避鱼类的免疫,也为哺乳动物抵抗细菌入侵提供了新的视角。